Dual Pathways to One Receptor: Structural Mechanisms of Ghrelin and Ibutamoren at GHSR
Blog 14
Abstract
The ghrelin receptor (GHSR), a class A G protein-coupled receptor, plays a central role in appetite regulation, energy balance, and growth hormone secretion. Despite its clinical relevance, the structural basis of GHSR activation by endogenous and synthetic ligands has remained elusive. In this study, high-resolution cryo-EM structures of GHSR bound to ghrelin and the non-peptidic agonist ibutamoren, each coupled to the Gi protein, reveal a shared dual-cavity binding mechanism. Ghrelin’s octanoylated N-terminus and ibutamoren’s hydrophobic moiety engage conserved receptor residues, initiating activation through a salt bridge and aromatic switch motif. Additionally, structural comparisons expose conformational diversity in Gi coupling, suggesting receptor flexibility may underlie signaling specificity. These insights offer a detailed blueprint for rational drug design targeting GHSR, including biased agonists for metabolic and endocrine disorders.
Introduction: From Hunger Hormone to Therapeutic Target
Ghrelin, a 28-amino acid peptide hormone primarily secreted by the stomach, plays a pivotal role in regulating energy homeostasis, appetite stimulation, and growth hormone (GH) secretion. Widely recognized as the “hunger hormone,” ghrelin acts via the growth hormone secretagogue receptor (GHSR)—a class A G protein-coupled receptor (GPCR)—to coordinate metabolic, endocrine, and neurobehavioral responses under physiological and pathological conditions.
Beyond its orexigenic effects, ghrelin modulates a range of central nervous system processes, including stress adaptation and reward signaling, implicating GHSR in psychiatric and neurodegenerative disorders. This broad functional spectrum has established GHSR as a high-value therapeutic target. Several GHSR agonists and antagonists are currently in preclinical and clinical development for diverse indications such as cachexia, gastroparesis, growth hormone deficiency, and substance use disorders. Among them, ibutamoren (also known as MK-0677 or LUM-201)—a potent, non-peptide, orally bioavailable GHSR agonist-has emerged as a lead candidate for GH stimulation in pediatric and adult populations.
Despite intense pharmacological interest, the structural underpinnings of ligand recognition and receptor activation for both endogenous (ghrelin) and synthetic agonists have remained incompletely understood. In a recent study published in Nature Communications, Liu et al. report high-resolution cryo-electron microscopy (cryo-EM) structures of the human GHSR in complex with ghrelin and ibutamoren, each bound to the heterotrimeric Gi protein. These structures provide critical insights into the molecular basis of GHSR activation, offering a framework for rational drug design and biased signaling modulation in metabolic and endocrine disorders.
Visualizing Activation: Cryo-EM Maps the Ghrelin-GHSR Interface
To elucidate the molecular basis of GHSR activation, the authors resolved two cryo-EM structures of the full-length human GHSR in complex with the Gi heterotrimer and either ghrelin or ibutamoren. Both complexes were determined at an overall resolution of 2.7 Å, enabling precise modeling of ligand–receptor interactions and conformational rearrangements associated with activation.
A defining feature of these structures is the dual-cavity ligand-binding pocket of GHSR. Ghrelin adopts an extended conformation, with its N-terminal peptide segment (Gly¹–Val¹²) inserted deep into cavity I, while its essential octanoyl group at Ser³ extends into cavity II. This acylation-dependent insertion into a hydrophobic cleft formed between transmembrane helices TM4, TM5, and TM6 is critical for receptor activation-a finding consistent with previous mutagenesis data demonstrating the functional necessity of Ser³ octanoylation.
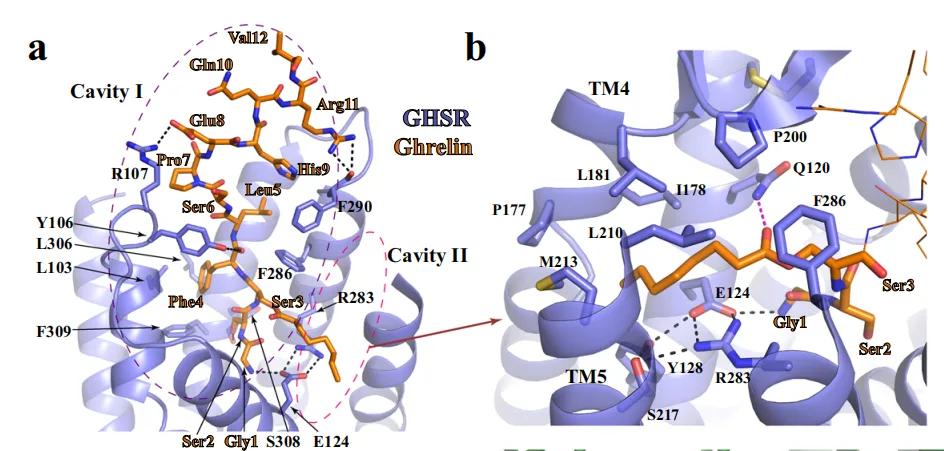
Ibutamoren, though structurally distinct and non-peptidic, mimics the spatial arrangement of ghrelin’s N-terminal residues. Its three molecular arms project into discrete regions of the binding pocket, effectively recapitulating ghrelin’s interactions in both cavities. Notably, one arm of ibutamoren occupies the same hydrophobic sub-pocket engaged by the ghrelin octanoyl group, establishing interactions with conserved residues such as I1784.60, L1814.63, and L2105.36. Additionally, ibutamoren forms hydrogen bonds with Q1203.29 and R2836.55, further stabilizing its binding.
These findings highlight a shared activation mechanism across chemically distinct ligands, wherein structural mimicry enables both peptide and small-molecule agonists to trigger conformational changes in GHSR. The data also underscore the plasticity of the GHSR binding site—a key consideration for structure-based drug discovery targeting this receptor.
Molecular Mechanism: How Ghrelin and Ibutamoren Unlock GHSR
The structural data reveal that agonist binding to GHSR induces a cascade of conformational rearrangements characteristic of GPCR activation, but with unique features that distinguish GHSR from canonical class A GPCRs.
Central to the activation mechanism is the E1243.33–R2836.55 salt bridge, which demarcates the boundary between cavities I and II. While present in both the active and inactive states, this salt bridge undergoes subtle positional shifts upon ligand engagement. In the active structures, ghrelin’s octanoyl group or ibutamoren’s phenyl moiety displaces R2836.55, forcing it toward a conserved aromatic cluster comprising W2766.48, F2796.51, H2806.52, and F3127.42. This structural motif—reminiscent of the “transmission switch” observed in other GPCRs—serves as a critical relay, propagating ligand-induced perturbations into global receptor activation.
Notably, W2766.48 adopts a toggled conformation, triggering the outward displacement of TM6 and repositioning of F2726.44, both hallmarks of the active GPCR state. Interestingly, TM7 exhibits an extended helical segment and an inward movement toward TM6—a rare configuration among class A GPCRs. This displacement appears to be a receptor-intrinsic feature rather than a direct result of ligand binding, as supported by mutagenesis experiments showing minimal impact on activity when the extracellular end of TM7 is altered.
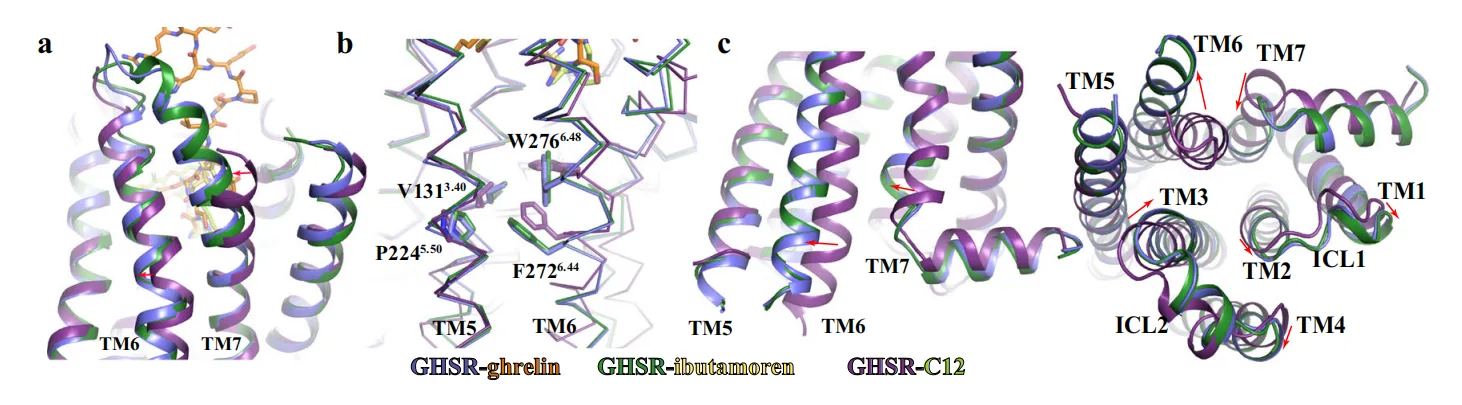
These collective shifts create a reorganized intracellular cavity that accommodates and stabilizes the heterotrimeric Gi protein. The unique choreography of TM6 and TM7, together with the aromatic cluster acting as a conformational fulcrum, defines a distinctive activation mechanism for GHSR, tailored to its specialized role in integrating metabolic and endocrine signals.
Coupling Specificity: Gi Protein Engagement and Conformational Diversity
Ligand binding and receptor activation culminate in the recruitment and engagement of intracellular transducers—in this case, the Gi heterotrimeric protein complex. Both ghrelin- and ibutamoren-bound GHSR structures revealed a near-identical Gi-coupling interface, indicating that distinct agonists can converge on a shared signaling output.
At the core of this interaction is the C-terminal α5 helix of Gαi, which inserts deeply into the cytoplasmic cleft formed by outwardly displaced TM6 and inwardly repositioned TM7. The hydrophobic interface is dominated by Gαi residues I344, L348, and L353, which interact with I1453.54, I2355.61, I2395.65, and L2656.37 on GHSR. Complementing these contacts, polar interactions are established between N347 and D350 of Gαi and A144 (backbone carbonyl) and K329 of GHSR. Additionally, R1413.50, a highly conserved residue in the DRY motif, forms a stabilizing hydrogen bond with C351 of Gαi, anchoring the complex.
The intracellular loop 2 (ICL2) further contributes to Gi engagement via interactions with F336, I343, I344 (Gαi) and P148, I149 (GHSR), forming a secondary hydrophobic interface. Interestingly, Gβ subunits also establish direct polar contacts with GHSR, reminiscent of what has been observed in other peptide GPCR-Gi complexes such as formylpeptide receptor 2 (FPR2), suggesting a shared mechanism for Gβ-mediated stabilization.
Strikingly, the authors identified two conformational states of the GHSR-Gi complex: a predominant Conformer 1, and a minor Conformer 2, resolved at 3.5 Å. The latter displays a ~15° rotation of the αN helix and a ~10° shift in α5 of Gαi, relative to the receptor. Despite this repositioning, the receptor’s intracellular conformation and Gi-binding interface remain largely unaltered. This finding is consistent with structural heterogeneity observed in the NTSR1-Gi complex, implying a highly dynamic coupling interface that may accommodate signaling bias or modulate effector specificity.
These results not only reinforce the canonical mode of Gi engagement in class A GPCRs but also highlight GHSR’s conformational flexibility—a feature that could be exploited for fine-tuning signaling outcomes via biased agonism.
Future Horizons: Structural Insights Fuel Rational Drug Design
This structural dissection of GHSR signaling provides a transformative framework for understanding how chemically distinct ligands—ghrelin and ibutamoren—achieve convergent receptor activation. By revealing the shared use of a dual-cavity binding pocket, the dynamic salt bridge–aromatic cluster relay, and the conformational plasticity in Gi coupling, this work sharpens our mechanistic view of GHSR function.
From a translational perspective, these findings open new avenues for rational design of GHSR-targeting therapeutics. The high-resolution binding modes offer a blueprint for engineering biased agonists or allosteric modulators capable of selectively engaging downstream signaling pathways—particularly relevant for treating disorders like cachexia, growth hormone deficiency, and metabolic syndromes.
Moreover, the structural flexibility observed in Gi coupling suggests a potential mechanism for ligand-dependent signaling bias, an area of increasing interest in GPCR pharmacology. Future studies may build on these insights to explore how alternative ligands modulate GHSR conformational ensembles and cellular outcomes.
Ultimately, this study underscores the power of cryo-EM–guided receptor pharmacology and affirms GHSR as a structurally accessible and druggable target within the GPCR superfamily.