Massively Parallel Nanopore Sensing: A New Era for Peptide Profiling and Protein Identification
Blog 249
Proteomics is undergoing a profound transformation. While mass spectrometry has long been the gold standard for protein analysis, researchers continue to face persistent challenges: resolving isomeric peptides, detecting subtle post-translational modifications (PTMs), distinguishing single amino acid variants (SAAVs), and validating antibody specificity with precision. As biological research increasingly demands peptide-level resolution, new technologies are emerging to address these limitations.
Among them, nanopore-based single-molecule sensing is rapidly gaining attention. A recently reported massively parallel nanopore platform integrates streamlined peptide chemistry, motor-driven translocation, and AI-powered analytics to enable high-throughput peptide profiling and protein identification. This development signals a major step toward scalable, sequence-sensitive peptide analytics.
Engineering a Peptide-Compatible Nanopore Workflow
A central innovation of this platform lies in its peptide library preparation strategy. Traditional nanopore systems were optimized for nucleic acids, not peptides. To bridge this gap, researchers developed an Oligo–Peptide–Oligo (OPO) conjugation strategy that enables peptides to be sequenced within a DNA-compatible framework.
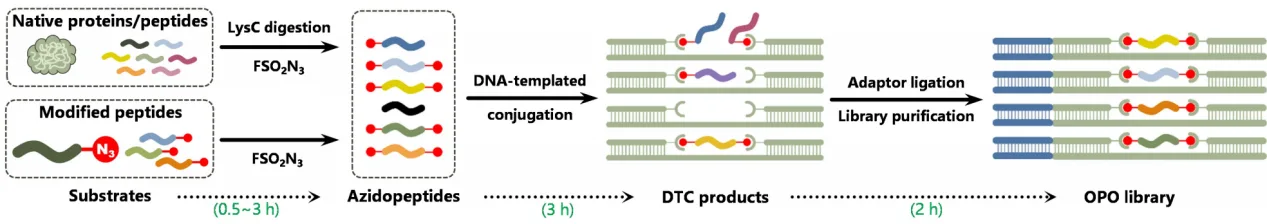
Step 1: LysC Digestion and Mild Azide Functionalization
Proteins are digested using LysC endopeptidase, generating peptides with C-terminal lysine residues. These peptides undergo mild azide modification using fluorosulfuryl azide (FSO₂N₃), which converts primary amines into azides under gentle conditions. This chemistry preserves peptide integrity and is compatible with native sequences and many PTMs.
Step 2: DNA-Templated Conjugation (DTC)
The azide-modified peptides are then assembled into OPO constructs through DNA-templated conjugation. Two DNA strands flank the peptide, enabling controlled translocation through a CsgG nanopore driven by a helicase motor. Importantly, abasic (AP) sites embedded in the DNA act as electrical signal markers, allowing automated extraction of the peptide-specific signal window.

Step 3: High-Throughput Parallel Sensing
Unlike early single-channel nanopore experiments, this platform uses a 256-channel array. Each run can generate over 100,000 single-molecule reads within two hours. Peptide translocation is monitored under optimized ionic conditions, and signal features such as:
- Current blockade ratio (I/I0)
- Dwell time (τoff)
- Signal variance
are extracted for downstream analysis.
Notably, peptide molar volume correlates strongly with blockade characteristics, reflecting how peptides move through the pore in a compact “blob-like” conformation. These measurable physical properties provide a foundation for peptide discrimination.
For researchers developing peptide standards, digestion fragments, or modified peptides, such structured library preparation underscores the importance of sequence-defined, high-purity synthetic peptides in next-generation proteomics workflows.
AI Transforms Stochastic Signals into Peptide Fingerprints
One of the major barriers in nanopore peptide sensing is signal variability. Peptides may enter the pore from either terminus, adopt flexible conformations, and produce overlapping electrical signatures. Traditional single-metric analysis (such as blockade depth alone) lacks sufficient resolution.
To overcome this, researchers implemented a two-stage analytical framework combining:
- Convolutional Neural Networks (CNNs)
- Density Matrix (DM) validation
CNN Classification
Each peptide read is normalized and downsampled into a standardized time-series format. A deep CNN architecture then classifies peptide identities based on temporal current patterns. Initial classification accuracy reached approximately 97.5%.
Density Matrix (DM) Reconfirmation
To further refine predictions, each peptide’s temporal signature is transformed into a 2D density matrix representing the statistical distribution of signal amplitudes across the event timeline. Reads are retained only if their patterns closely match the reference DM.
This CNN-DM integration increased classification accuracy to over 99%.
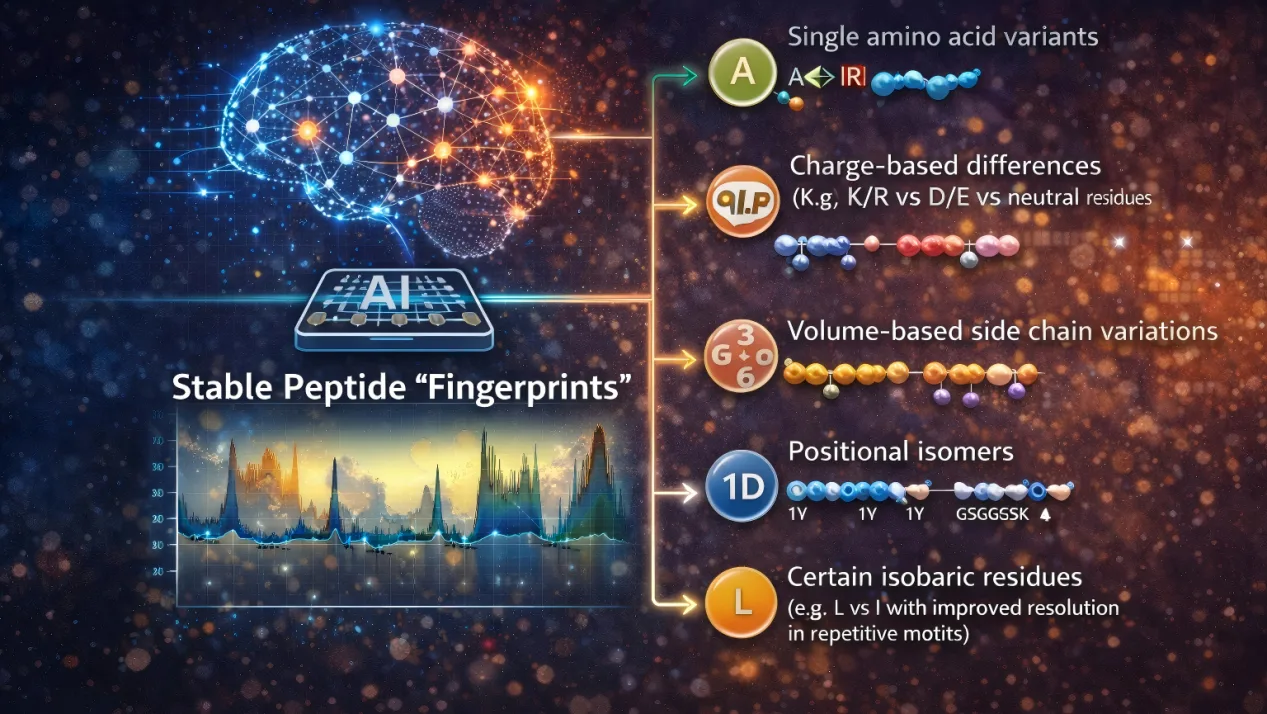
The result is transformative: noisy single-molecule electrical traces become stable peptide “fingerprints.” This approach enables reliable discrimination of:
- Single amino acid variants (SAAVs)
- Charge-based differences (e.g., K/R vs D/E vs neutral residues)
- Volume-based side chain variations
- Positional isomers
- Certain isobaric residues (e.g., L vs I with improved resolution in repetitive motifs)
For laboratories designing mutation libraries or isomeric peptide panels, such resolution highlights the importance of precisely synthesized sequence variants for AI model training and validation.
Sensitive Detection of Post-Translational Modifications
PTMs represent one of the most critical dimensions of functional proteomics. This nanopore platform demonstrates strong sensitivity to physicochemical changes introduced by modifications.
High-Resolution PTMs
Modifications that significantly alter charge or volume—such as phosphorylation or lipidation—were classified with high accuracy. Even dual phosphorylation events on specific residues could be distinguished.
Naturally occurring modifications were also successfully detected, including:
- Deamidation (Q → E substitution)
- Single and dual phosphorylation events
More Subtle Modifications
Modifications such as methylation or acetylation, which introduce smaller structural changes, were more challenging but still detectable at moderate accuracy levels.
These findings indicate that nanopore sensing combined with AI can directly detect PTMs without specialized enrichment tags or affinity reagents. For researchers developing modified peptides—phosphorylated, acetylated, methylated, lipidated—such analytical compatibility is particularly valuable.
Redefining Antibody Validation Through Peptide Profiling
Antibody validation remains a persistent bottleneck in proteomics. Commercial antibodies often lack detailed epitope mapping data, and batch variability can compromise reproducibility.
This nanopore system introduces a streamlined workflow for antibody characterization.
Epitope Mapping with Overlapping Peptides
Using six overlapping fragments of human C-peptide, researchers:
- Generated OPO libraries for each fragment
- Pooled them equally
- Incubated with antibody-coated magnetic beads
- Quantified enrichment via nanopore read distribution
Distinct antibodies showed different enrichment patterns, revealing epitope preferences. In some cases, nanopore analysis contradicted manufacturer claims about antibody pairing suitability.
Semi-Quantitative Affinity Profiling
Using the FLAG-tag system and four point mutants, researchers observed strong correlation between nanopore read abundance and known binding affinities. This demonstrates that the platform provides not only epitope mapping but also semi-quantitative affinity assessment.
For companies and researchers developing:
- Overlapping epitope peptides
- Mutant peptide libraries
- Affinity-optimized antigen variants
this approach represents a rapid, cost-effective complement to traditional ELISA, SPR, or LC-MS validation methods.
From Peptide Fingerprints to Protein Identification
Beyond peptide-level discrimination, the system enables protein identification through peptide fingerprinting.
The workflow is straightforward:
- Digest proteins with LysC
- Generate OPO libraries
- Sequence peptides in parallel
- Classify reads against trained CNN-DM reference libraries
- Compile peptide signatures
In a blinded experiment, three proteins were successfully identified from their enzymatic digests. Multi-peptide signatures proved sufficient for robust protein-level assignment, even when minor classification artifacts were present.
This strategy demonstrates that full sequence coverage is not always required. Instead, statistically robust multi-peptide fingerprints can support accurate protein identification.
For proteomics researchers, this suggests a future where targeted peptide panels—rather than exhaustive proteome coverage—enable efficient biomarker discovery and diagnostic profiling.
The Future of AI-Driven Peptide Analytics
Massively parallel nanopore sensing marks a pivotal shift in proteomics. By integrating mild peptide chemistry, scalable hardware, and deep learning analytics, this platform transforms peptides into quantifiable, high-dimensional electrical fingerprints.
Key implications include:
- High-throughput PTM detection
- Precise discrimination of subtle sequence variants
- Rapid antibody epitope validation
- Semi-quantitative affinity profiling
- Multi-peptide protein identification
As compatibility with commercial nanopore sequencers improves, and as AI models are trained on larger peptide libraries, the potential for clinical diagnostics and biomarker screening becomes increasingly tangible.
At the foundation of this technological evolution lies a simple but essential requirement: high-quality, customizable peptide libraries. Defined sequences, positional variants, and site-specific modifications are critical for building accurate analytical models and reference datasets.
The convergence of peptide chemistry and AI-driven nanopore sensing is reshaping how researchers profile, validate, and identify proteins—one peptide at a time.
Reference
Wang, J., Chen, J., Pan, H., Luo, F., Qin, W., Zeng, H., … & Xu, X. (2026). Nanopore-based massively parallel sensing for peptide profiling and protein identification. Nature Communications.https://doi.org/10.1038/s41467-026-69628-1
Cressiot, B., Bacri, L., & Pelta, J. (2020). The promise of nanopore technology: advances in the discrimination of protein sequences and chemical modifications. Small Methods, 4(11), 2000090.https://doi.org/10.1002/smtd.202000090
Bakshloo, M. A., Yahiaoui, S., Piguet, F., Pastoriza-Gallego, M., Daniel, R., Mathé, J., … & Oukhaled, A. (2022). Polypeptide analysis for nanopore-based protein identification. Nano Research, 15(11), 9831-9842.https://doi.org/10.1007/s12274-022-4610-1
Zhang, M., Tang, C., Wang, Z., Chen, S., Zhang, D., Li, K., … & Geng, J. (2024). Real-time detection of 20 amino acids and discrimination of pathologically relevant peptides with functionalized nanopore. Nature Methods, 21(4), 609-618.https://doi.org/10.1038/s41592-024-02208-7