Next-Generation PROTACs: Peptide Strategies for Advanced Therapeutics
Blog 150
Abstract
Proteolysis-targeting chimeras (PROTACs) have emerged as a transformative modality in drug discovery by leveraging the ubiquitin–proteasome system to selectively degrade disease-associated proteins. While small-molecule PROTACs have demonstrated success, their reliance on well-defined ligandable pockets leaves many clinically relevant proteins beyond reach. Peptide-based PROTACs (p-PROTACs) address this limitation by recognizing large protein–protein interaction (PPI) surfaces, thereby expanding the degradable proteome. This article examines the mechanistic basis of peptide PROTACs, their distinct advantages in terms of specificity, design flexibility, and target scope, as well as the pharmacological challenges of stability, permeability, and delivery. Emerging strategies—including chemical modifications, cell-penetrating conjugates, and nanoparticle carriers—are discussed as potential solutions. Finally, the review highlights promising applications in oncology, neurodegeneration, and infectious disease, and considers future directions driven by advances in structural biology, computational modeling, and delivery science. Peptide PROTACs thus represent a powerful and versatile tool for next-generation therapeutics.
Introduction: Redefining the Scope of Drug Discovery
In recent years, proteolysis-targeting chimeras (PROTACs) have emerged as a transformative modality in drug discovery, offering a paradigm shift from traditional small-molecule inhibition. Rather than merely blocking enzymatic activity, PROTACs harness the ubiquitin–proteasome system to induce the selective degradation of disease-associated proteins. This catalytic mechanism not only ensures complete removal of pathogenic proteins but also enables sustained pharmacological effects at substoichiometric doses, distinguishing PROTACs from conventional therapeutic strategies.
However, the scope of small-molecule PROTACs remains restricted. Their success depends on the availability of high-affinity ligands capable of engaging well-defined binding pockets. As a result, many critical proteins—such as transcription factors, scaffolding molecules, and key signaling mediators—remain largely “undruggable.” This limitation has spurred growing interest in peptide-based PROTACs (p-PROTACs) as an innovative alternative.
Peptides are uniquely suited to recognize broad, flat protein–protein interaction (PPI) surfaces that small molecules typically cannot access. By exploiting this capability, peptide PROTACs markedly expand the degradable proteome and open new therapeutic possibilities across oncology, neurology, and infectious disease. This blog will explore the mechanistic principles, advantages, current challenges, and future directions of peptide PROTACs, underscoring their potential to redefine the frontiers of modern drug discovery.
Mechanistic Framework of Peptide PROTACs
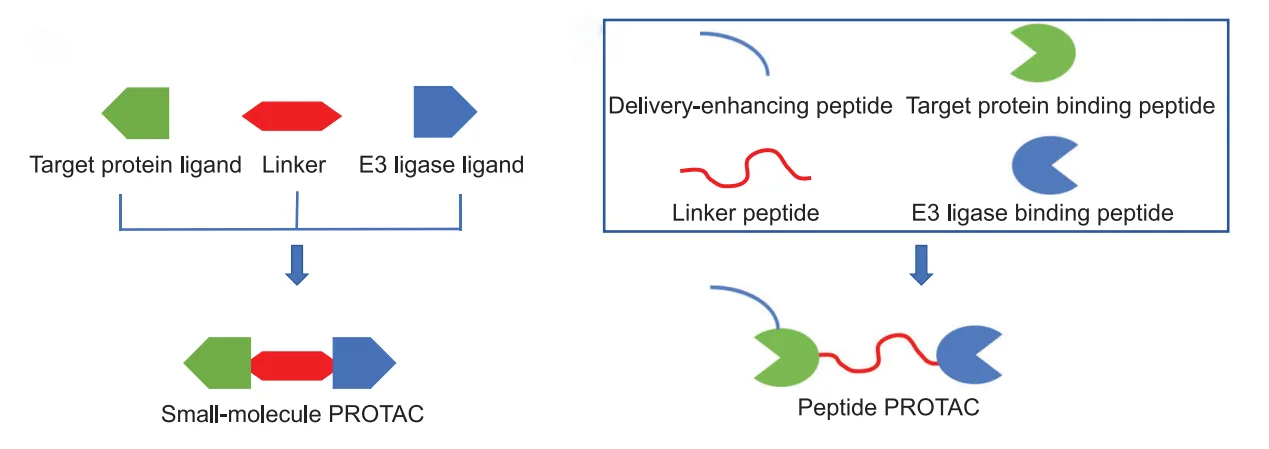
The conceptual framework of peptide PROTACs (p-PROTACs) mirrors that of their small-molecule counterparts, yet their structural composition imparts distinct advantages. A typical p-PROTAC consists of three essential modules: (1) a target-recognizing peptide ligand, (2) a linker region that provides spatial flexibility, and (3) a motif or ligand that recruits an E3 ubiquitin ligase. Once introduced into the cellular environment, the bifunctional construct simultaneously engages the protein of interest and the E3 ligase, thereby promoting polyubiquitination of the target and directing it toward proteasomal degradation.
Unlike small molecules, peptides can be designed to recognize extended protein–protein interaction (PPI) surfaces. Many disease-relevant proteins, such as transcription factors or scaffolding proteins, lack deep hydrophobic pockets suitable for small-molecule binding. By exploiting amino acid sequences or secondary structural elements, peptides achieve high binding specificity to these otherwise intractable targets. This capacity effectively broadens the “druggable proteome,” positioning peptide PROTACs as a powerful complement to existing modalities.
Moreover, the modularity of peptides enables rapid design and optimization. Advances in solid-phase peptide synthesis (SPPS) and combinatorial peptide libraries facilitate the systematic identification of high-affinity ligands. In parallel, structural biology and computational modeling contribute to the rational assembly of peptide–ligase linkages with optimal geometry. Collectively, these innovations underscore how p-PROTACs harness the precision of peptide recognition while maintaining the catalytic degradation principle central to the PROTAC strategy.
Distinct Advantages of the Peptide PROTAC Approach
The appeal of peptide-based PROTACs lies in their capacity to overcome limitations that constrain small-molecule degraders. Perhaps the most significant advantage is binding specificity. Peptides are inherently well suited to engage large and relatively flat protein–protein interaction (PPI) interfaces, which are inaccessible to conventional ligands. By recapitulating natural binding motifs or engineered sequences, peptide PROTACs achieve high affinity and selectivity, thereby minimizing off-target interactions that often compromise small-molecule approaches.
A second advantage is structural flexibility and tunability. Peptides can be readily modified to enhance stability and activity. Chemical strategies such as cyclization, hydrocarbon stapling, incorporation of D-amino acids, and N-methylation improve conformational rigidity, resistance to proteolytic degradation, and membrane permeability. These modifications allow researchers to fine-tune pharmacological profiles without sacrificing binding fidelity.
Third, peptide PROTACs benefit from rapid discovery and optimization pipelines. The maturation of solid-phase peptide synthesis (SPPS) and high-throughput peptide libraries enables efficient screening of candidate sequences. In addition, advances in computational modeling and structural biology allow rational design of peptides that mimic or disrupt critical PPIs, accelerating the identification of effective target ligands.
From a therapeutic perspective, peptide PROTACs significantly expand the range of druggable targets. While small molecules are often restricted to kinases or enzymes with defined pockets, peptides can address transcription factors, adaptors, and signaling hubs—classes of proteins long considered “undruggable.” This capacity is especially valuable in fields such as oncology and neurodegeneration, where pathogenic proteins frequently lack enzymatic activity but exert their effects through multiprotein complexes.
Taken together, these advantages position peptide PROTACs as a versatile and innovative platform, offering precision, adaptability, and expanded therapeutic reach that complements and extends the scope of traditional small-molecule PROTACs.
Pharmacological Challenges and Emerging Solutions
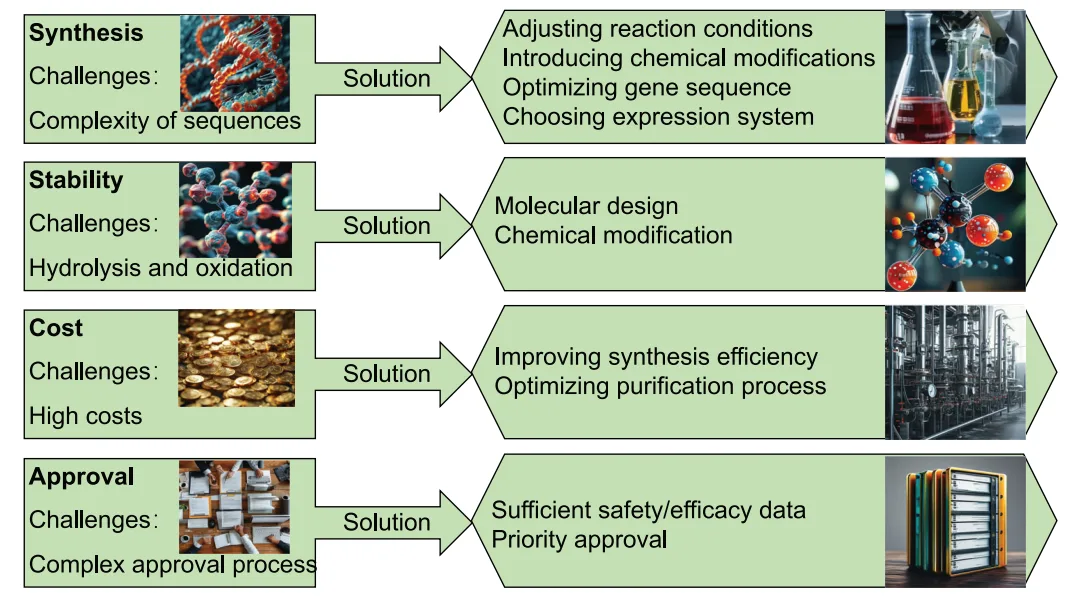
Despite their promise, peptide PROTACs face considerable barriers to clinical translation. The most immediate challenge is poor cell permeability. Due to their relatively large molecular size and polar backbones, peptides often exhibit limited capacity to traverse the plasma membrane, restricting their intracellular bioavailability. This obstacle is compounded by their inherent susceptibility to proteolytic degradation, which shortens half-life and diminishes systemic stability.
A second challenge lies in delivery. While small molecules generally display favorable pharmacokinetics, peptides require specialized strategies to reach therapeutic concentrations in target tissues. This issue is particularly pronounced for diseases affecting the central nervous system, where the blood–brain barrier (BBB) poses an additional hurdle.
Several approaches are under investigation to mitigate these limitations. Chemical modifications—including cyclization, hydrocarbon stapling, and the incorporation of D-amino acids—have been shown to enhance metabolic stability and improve membrane penetration. Similarly, PEGylation and lipidation can prolong circulation time and modulate biodistribution.
In parallel, delivery technologies are rapidly evolving. Conjugation to cell-penetrating peptides (CPPs) has demonstrated success in facilitating cytosolic entry, while encapsulation within nanoparticles, liposomes, or polymeric carriers provides protection from enzymatic degradation and enables controlled release. In preclinical studies, these delivery vehicles have enhanced intracellular accumulation of peptide PROTACs and improved target engagement.
Another promising strategy involves the design of tissue-homing peptides, which confer organ- or cell-type specificity, thereby reducing systemic exposure and potential off-target effects.
Collectively, these innovations suggest that while pharmacokinetic and delivery challenges remain substantial, they are surmountable through rational chemical engineering and advanced formulation approaches. The convergence of peptide chemistry, nanotechnology, and drug delivery science will be critical in unlocking the therapeutic potential of peptide PROTACs.
Therapeutic Applications and Future Directions
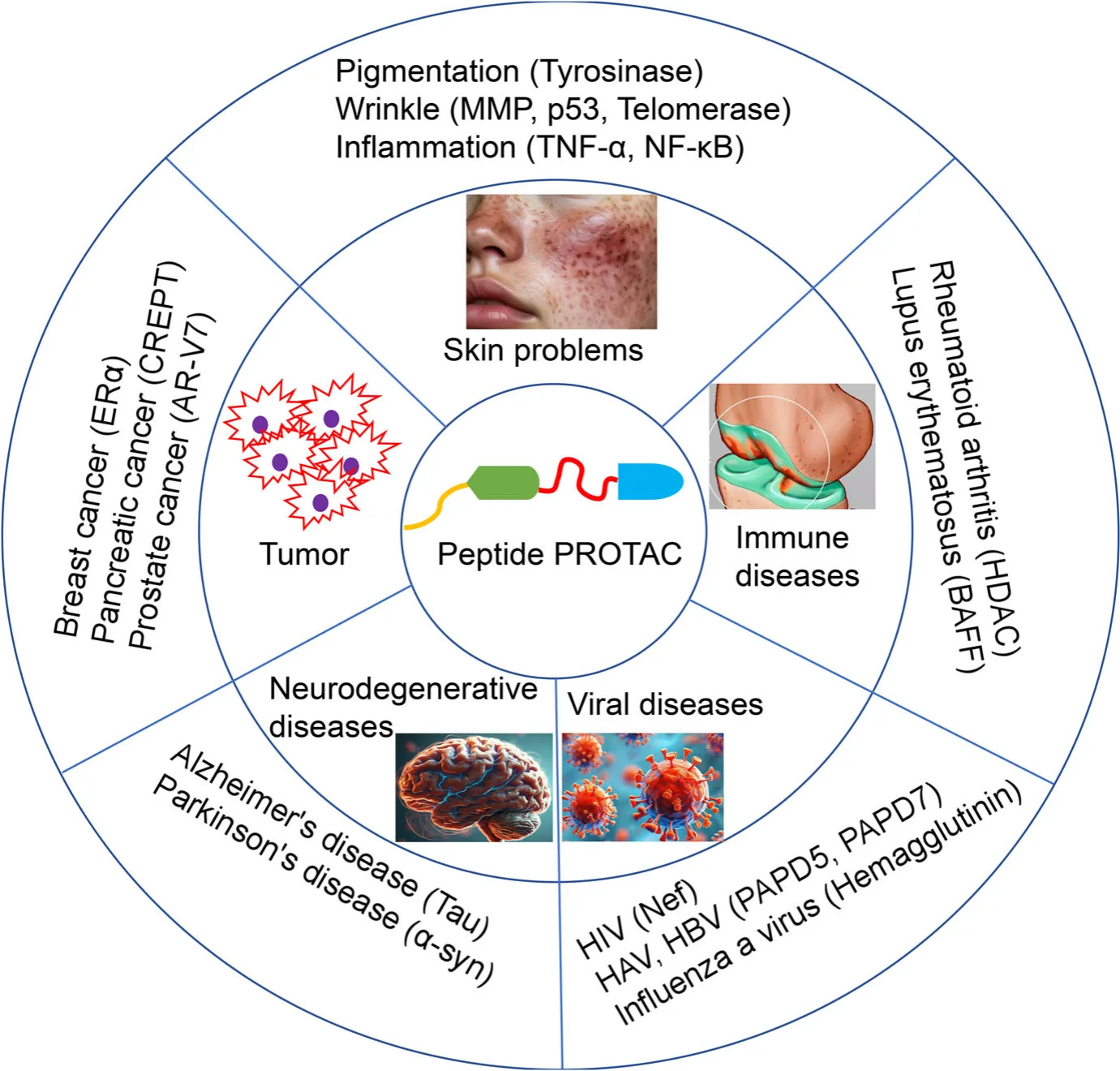
The therapeutic potential of peptide PROTACs is rapidly gaining attention, with early studies demonstrating proof-of-concept across diverse disease areas. In oncology, peptide PROTACs have been engineered to degrade transcription factors such as c-Myc and STAT3, long recognized as central oncogenic drivers yet historically inaccessible to small-molecule inhibitors. By dismantling these master regulators, peptide PROTACs provide a new means of disrupting malignant signaling networks.
In the realm of neurodegeneration, peptide-based degraders targeting α-synuclein and tau represent promising strategies to address protein aggregation disorders such as Parkinson’s and Alzheimer’s disease. Traditional inhibitors are ineffective against these intrinsically disordered proteins, but peptide PROTACs offer a route to their selective clearance. Similarly, in infectious disease, peptide PROTACs have been designed to target viral proteins that lack conventional ligand-binding pockets, highlighting their versatility in combating pathogens.
Looking forward, the field is poised for acceleration through integration with emerging technologies. Artificial intelligence and machine learning are increasingly applied to peptide design, enabling rapid prediction of binding motifs and optimization of physicochemical properties. Advances in structural biology, particularly cryo-electron microscopy and computational docking, will further refine rational design strategies. At the same time, progress in nanoparticle-based delivery systems and cell-penetrating carriers is expected to address pharmacokinetic limitations, enhancing translational feasibility.
Collectively, these developments suggest that peptide PROTACs are positioned to redefine the boundaries of drug discovery. By expanding the degradable proteome and enabling precise modulation of previously inaccessible targets, they represent a transformative tool for next-generation therapeutics. While significant challenges remain, the convergence of peptide chemistry, structural biology, and delivery science offers a clear trajectory toward clinical application and broader adoption in biomedical research.
Reference
Zhu, Y., Dai, Y., & Tian, Y. (2025). The Peptide PROTAC Modality: A New Strategy for Drug Discovery. MedComm, 6(4), e70133.
https://doi.org/10.1002/mco2.70133
Sakamoto, K. M., Kim, K. B., Kumagai, A., Mercurio, F., Crews, C. M., & Deshaies, R. J. (2001). Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proceedings of the National Academy of Sciences, 98(15), 8554-8559.
https://doi.org/10.1073/pnas.141230798
Sahni, A., Ritchey, J. L., Qian, Z., & Pei, D. (2024). Cell-penetrating peptides translocate across the plasma membrane by inducing vesicle budding and collapse. Journal of the American Chemical Society, 146(36), 25371-25382.
https://doi.org/10.1021/jacs.4c10533
Hariri, R., Saeedi, M., & Akbarzadeh, T. (2021). Naturally occurring and synthetic peptides: Efficient tyrosinase inhibitors. Journal of Peptide Science, 27(7), e3329.