Recent Advances in Peptide Linkers for Antibody-Drug Conjugates: Building Smarter ADCs for Precision Cancer Therapy
Antibody-drug conjugates (ADCs) have become one of the most important therapeutic platforms in modern oncology. By combining the high target specificity of monoclonal antibodies with the strong cytotoxic activity of small-molecule payloads, ADCs offer a way to deliver potent drugs directly to cancer cells while reducing systemic exposure. This “targeted delivery” concept has transformed the treatment landscape for several hematologic malignancies and solid tumors.
However, the clinical performance of an ADC does not depend only on the antibody or the payload. Between these two components lies a smaller but equally critical element: the linker. The linker determines how stably the payload remains attached during circulation and how efficiently it is released at the disease site. In many cases, the linker is the difference between a highly effective ADC and one with unacceptable toxicity or insufficient efficacy.
Among linker technologies, cleavable peptide linkers have become especially important. According to the reviewed article, 19 ADCs have been approved globally, and 16 of them use cleavable linkers. Of these, 11 approved ADCs use enzyme-cleavable peptide linkers, showing how central peptide chemistry has become in ADC design. In clinical-stage ADCs, peptide linkers such as Val-Cit, Val-Ala, and Gly-Gly-Phe-Gly are widely used because they can respond to tumor-associated or lysosomal proteases and release payloads selectively in target cells or the tumor microenvironment.
Why the Linker Matters in ADC Design
A typical ADC contains three main components: an antibody, a cytotoxic payload, and a linker. The antibody recognizes a tumor-associated antigen, the payload kills the cancer cell, and the linker connects the two. Although the linker may appear to be a simple bridge, it controls multiple properties that directly influence therapeutic outcome.
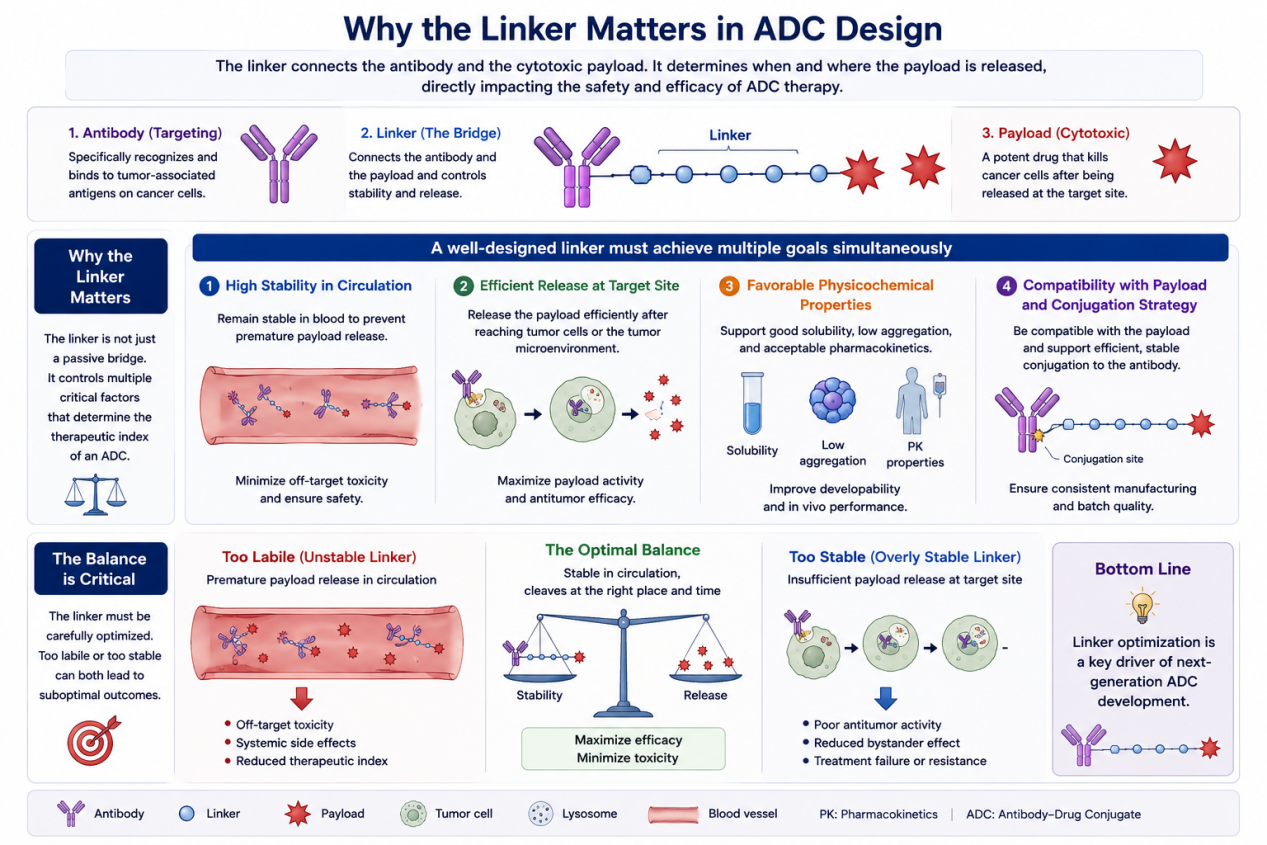
A well-designed ADC linker must achieve several goals at once. First, it must remain stable in systemic circulation to avoid premature payload release. Second, it must release the payload efficiently after the ADC reaches the tumor cell or tumor microenvironment. Third, it must support favorable physicochemical properties, including solubility, low aggregation, and acceptable pharmacokinetics. Fourth, it must be compatible with the chosen payload and conjugation strategy.
This balance is difficult. If the linker is too labile, the payload may be released into the bloodstream, leading to off-target toxicity. If the linker is too stable, the payload may not be released efficiently, reducing antitumor activity. This is why linker optimization has become a major focus in next-generation ADC development.
Cleavable vs. Noncleavable Linkers
ADCs can use either cleavable or noncleavable linkers. Noncleavable linkers remain attached to the payload until the antibody is degraded inside the target cell, typically within lysosomes. These linkers can provide strong plasma stability, but they often release charged amino acid-linker-payload metabolites. Such metabolites may have limited membrane permeability and reduced bystander killing effects, making them more suitable for certain tumors with strong antigen expression.
Cleavable linkers, by contrast, are designed to respond to biological stimuli. These stimuli can include acidic pH, reducing conditions, or specific enzymes. Peptide linkers belong to the enzyme-cleavable category. They are designed to be recognized and cleaved by proteases such as cathepsins, legumain, plasmin, or other tumor-associated enzymes. This enables the ADC to release a free or active payload after internalization or within the tumor microenvironment.
The attached review emphasizes that peptide linkers are favored in next-generation ADC construction because they offer a strong balance between stability and responsiveness. They also provide modularity: by changing amino acid sequence, stereochemistry, spacer design, and hydrophilic modifications, researchers can tune linker stability, cleavage rate, selectivity, and compatibility with different payloads.
Val-Cit Linkers: The Classic ADC Peptide Linker
The Val-Cit, or valine-citrulline, linker is one of the most successful peptide linkers in ADC history. It is best known for its use in vedotin-type ADCs, where the Val-Cit-PABC linker connects the antibody to monomethyl auristatin E (MMAE). The PABC spacer is self-immolative: after protease cleavage of the peptide sequence, the spacer undergoes elimination to release the free payload.
Val-Cit linkers were originally designed to be sensitive to cathepsin B, a lysosomal cysteine protease. However, more recent research shows that cleavage is not limited to cathepsin B. Other cathepsins, including cathepsins L, S, and Z, may also contribute, depending on tumor type, target antigen, and intracellular trafficking route. Some evidence also suggests that linker cleavage may occur outside lysosomes, including in late or recycling endosomes.
The success of Val-Cit linkers is clear from clinical use. Several approved ADCs rely on the classical Val-Cit-PABC-MMAE format. However, this linker class also has limitations. One important problem is species-dependent instability, particularly in mouse plasma, where carboxylesterase 1C can cleave certain Val-Cit-containing constructs. This complicates preclinical evaluation because mouse stability may not accurately reflect human stability. Another concern is potential cleavage by human neutrophil elastase, which may contribute to off-target toxicity. These issues have driven the search for improved peptide sequences with better cross-species stability and protease selectivity.
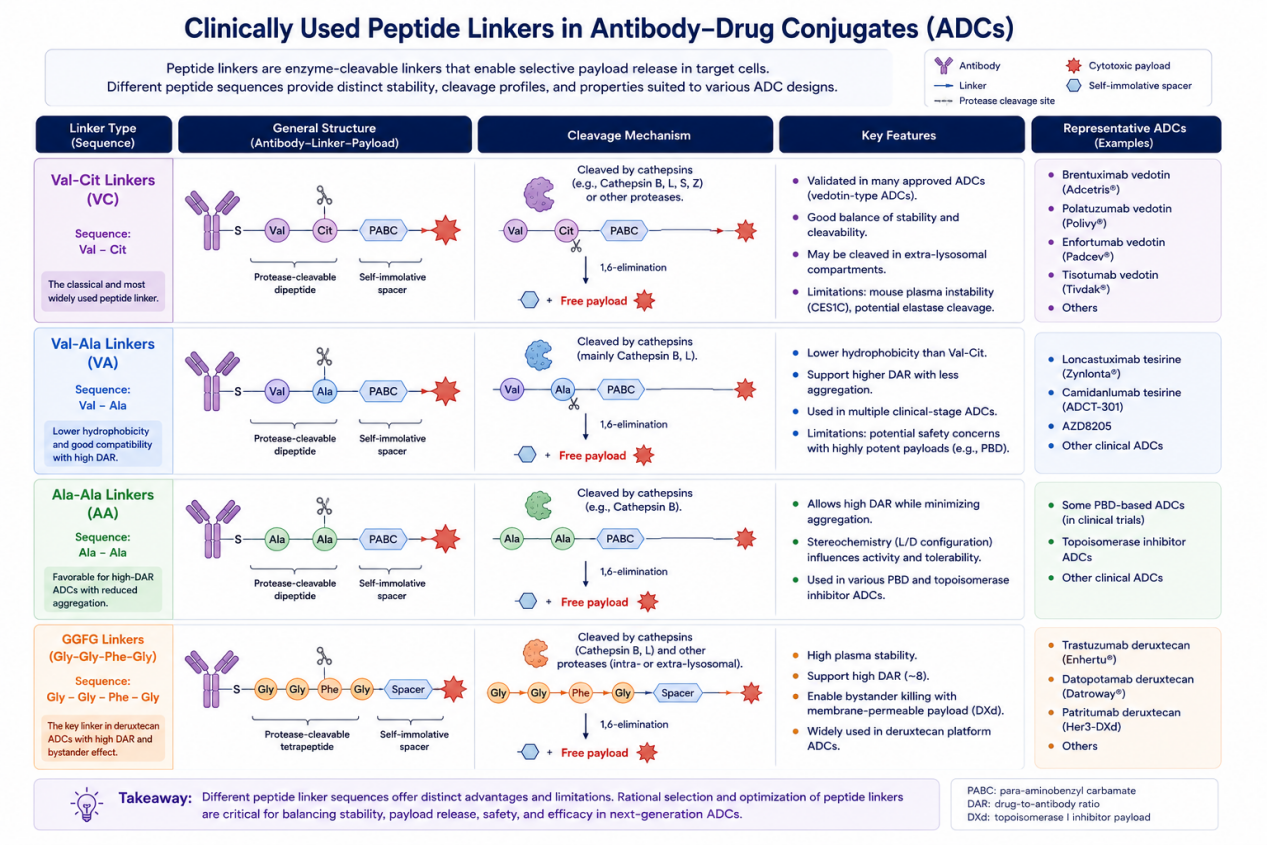
Val-Ala Linkers: Lower Hydrophobicity and Better ADC Properties
Val-Ala is another clinically important dipeptide linker. Like Val-Cit, it has been associated with cathepsin-mediated cleavage. Compared with Val-Cit, Val-Ala can offer lower hydrophobicity, which may improve conjugation behavior and reduce aggregation, especially when paired with hydrophobic payloads.
Early studies showed that Val-Ala linkers could support higher drug-to-antibody ratios while maintaining relatively low aggregation. This is important because high-DAR ADCs can deliver more payload per antibody, but they often suffer from poor solubility, faster clearance, and greater aggregation if the linker-payload is too hydrophobic.
Val-Ala linkers have been used in multiple clinical ADC formats, including PBD-based ADCs and topoisomerase inhibitor ADCs. Some Val-Ala ADC programs have faced safety challenges, especially when combined with highly potent PBD payloads. However, other Val-Ala-based ADCs continue to advance clinically, often with additional PEG or hydrophilic modifications to improve physicochemical behavior.
Ala-Ala Linkers: A Route Toward High-DAR ADCs
Ala-Ala linkers have gained attention because of their favorable compatibility with high-DAR ADC design. The attached review highlights research showing that Ala-Ala linkers can reduce aggregation compared with Val-Cit and Val-Ala in certain high-DAR constructs. This is highly relevant because next-generation ADCs increasingly aim to carry more payload while preserving stability and manufacturability.
The stereochemistry of Ala-Ala linkers also matters. Studies have shown that different L- and D-alanine configurations can influence cleavage, activity, and tolerability. For example, the L-Ala-L-Ala configuration has been associated with a strong therapeutic index in some ADC designs. Ala-Ala-Ala tripeptide linkers have also been explored, with stereochemical placement influencing cytotoxic activity.
These findings show that peptide linkers should not be treated as simple interchangeable sequences. Even small changes in amino acid configuration can affect enzyme recognition, intracellular release, payload exposure, and therapeutic window.
GGFG Linkers and the Deruxtecan Revolution
The Gly-Gly-Phe-Gly, or GGFG, tetrapeptide linker has become one of the most clinically influential peptide linkers due to its use in deruxtecan ADCs. Trastuzumab deruxtecan was the first approved ADC using the GGFG-DXd linker-payload system, and it has had major clinical impact, especially in HER2-positive and HER2-low breast cancer settings.
GGFG linkers provide strong systemic stability while enabling protease-mediated payload release. A key feature of deruxtecan ADCs is their ability to support a high DAR, commonly around 8, while maintaining acceptable properties. The linker-payload design also enables bystander killing, in which released membrane-permeable payload can affect neighboring tumor cells, including those with lower antigen expression.
The review notes that GGFG cleavage may involve not only cathepsin B but also cathepsin L and extracellular tumor proteases. This is important because it expands the possible mechanisms of payload release beyond classical lysosomal degradation. In some tumor contexts, extracellular cleavage in the tumor microenvironment may contribute significantly to efficacy.
The clinical success of trastuzumab deruxtecan, patritumab deruxtecan, datopotamab deruxtecan, and related ADCs demonstrates that tetrapeptide linkers can be highly effective when paired with suitable payloads, spacers, and antibody targets.
TMALIN and VKG Linkers: Activating Payload Release in the Tumor Microenvironment
A newer direction in peptide linker design is the development of linkers that can be activated in the tumor microenvironment rather than relying strictly on ADC internalization. The review discusses Val-Lys-Gly-based linkers and the TMALIN platform, which is designed to release payload through tumor microenvironment-associated enzymatic cleavage.
This strategy is attractive because some tumor targets may not internalize efficiently. Traditional ADCs often depend on antigen binding, internalization, trafficking to lysosomes, and proteolytic degradation. If any step is inefficient, payload release may be limited. A tumor microenvironment-activatable linker could expand the range of usable targets, including noninternalizing or poorly internalizing antigens.
MediLink’s TMALIN platform has generated several clinical ADC candidates targeting HER3, B7H3, CDH17, and VEGF. The VEGF-targeting ADC is especially notable because soluble VEGF is not a conventional internalizing cell-surface antigen. This suggests that extracellularly activatable linker technology may broaden ADC target space.
However, the precise proteases responsible for VKG linker cleavage remain incompletely defined. This highlights a broader challenge in linker development: many new linkers show promising activity, but their exact in vivo cleavage pathways require deeper mechanistic validation.
Next-Generation Peptide Linker Strategies
Although classical peptide linkers have enabled major ADC successes, they also present limitations. The field is now moving toward next-generation strategies that improve stability, selectivity, solubility, and therapeutic window.
1. Optimizing Peptide Sequences
One strategy is to modify peptide sequences to avoid unwanted cleavage while preserving tumor-associated activation. For example, EVCit and EGCit linkers were designed to improve stability and reduce cleavage by off-target enzymes. Introducing glutamic acid at the P3 position improved mouse plasma stability by reducing susceptibility to carboxylesterase-mediated cleavage. Further modification to EGCit helped resist human neutrophil elastase while maintaining antitumor activity.
This approach shows how rational sequence optimization can fine-tune protease selectivity. Instead of using one universal peptide linker, ADC developers can design linkers for specific enzymatic environments and safety requirements.
2. Legumain-Cleavable Linkers
Legumain has emerged as an alternative protease for ADC activation. Asn-containing linkers, such as Gly-Asn-Asn or Ala-Ala-Asn, can be cleaved by legumain and may offer high serum stability and resistance to neutrophil elastase. These linkers may be useful in tumors where legumain expression is high.
However, tumor protease expression varies across cancer types and even between patients. Therefore, legumain-cleavable ADCs may require careful patient selection or biomarker-based development.
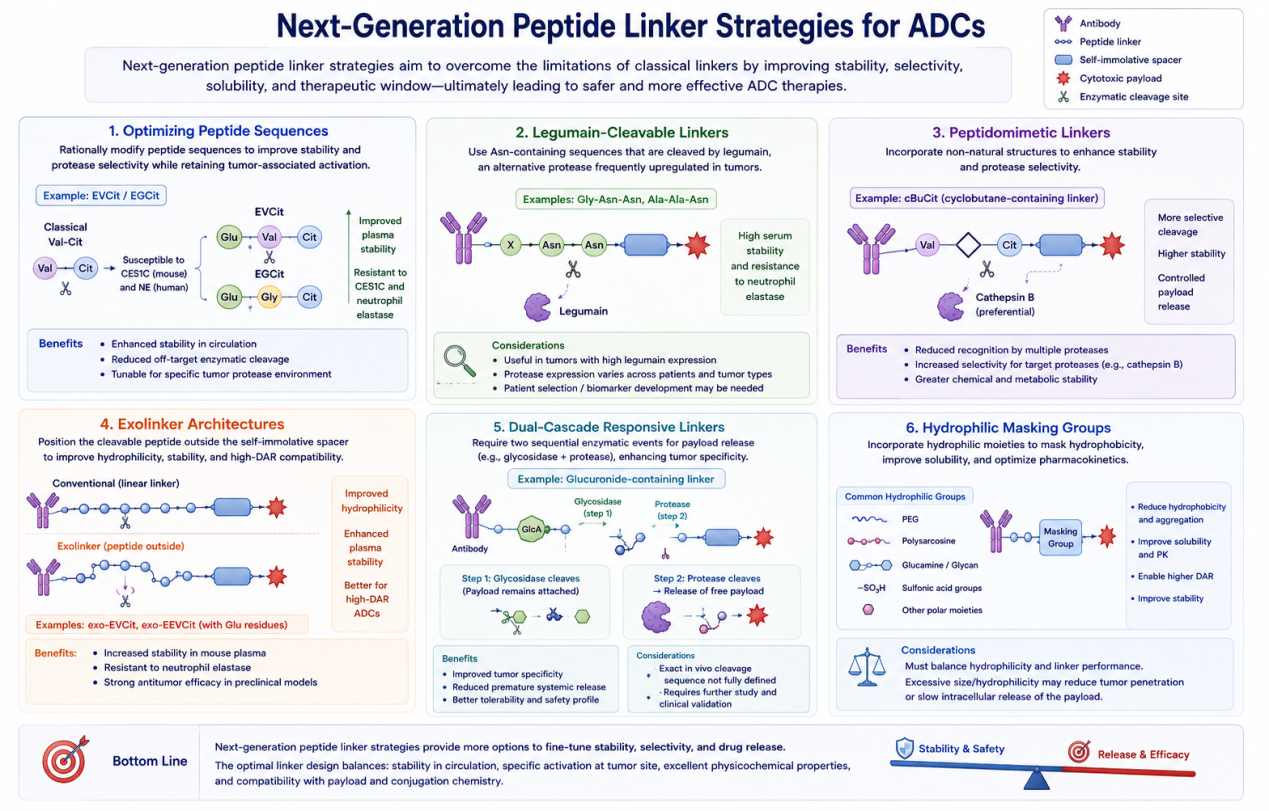
3. Peptidomimetic Linkers
Peptidomimetic linkers replace or modify parts of the peptide structure to improve stability and enzyme specificity. One example discussed in the review is cBuCit, a cyclobutane-containing linker designed to enhance cathepsin B selectivity. Peptidomimetic linkers may reduce unwanted cleavage while retaining controlled release.
This strategy is valuable because natural peptide sequences may be recognized by multiple proteases. Peptidomimetics can make the linker more selective and chemically robust.
4. Exolinker Architectures
The exolinker strategy changes the spatial positioning of the cleavable peptide relative to the self-immolative spacer. Instead of placing the peptide in a conventional linear arrangement, the cleavable sequence is positioned externally. This architecture can improve hydrophilicity, stability, and compatibility with high-DAR ADCs.
The review describes exo-EVCit and exo-EEVCit linkers, which combine exolinker design with hydrophilic glutamate residues. These linkers showed improved mouse plasma stability, resistance to neutrophil elastase, and strong antitumor efficacy in preclinical models.
5. Dual-Cascade Responsive Linkers
Dual-cascade responsive linkers require sequential activation by two enzymatic events. For example, glucuronide or glycoside groups can be incorporated into peptide linker structures, creating a linker that may require both glycosidase and protease activity for payload release.
This strategy can improve tumor specificity because two biological conditions must be met before the payload is released. It may reduce premature systemic release and improve tolerability. However, as the review notes, the exact cleavage sequence and in vivo validation of these cascade mechanisms still require further study.
6. Hydrophilic Masking Groups
Hydrophobic linker-payloads can cause ADC aggregation, rapid clearance, and limited high-DAR feasibility. Hydrophilic masking strategies address this problem by incorporating PEG, polysarcosine, glucamine, sugar groups, sulfonic acid groups, or other polar moieties.
PEGylated linkers have been shown to reduce hydrophobicity and improve pharmacokinetics. Polysarcosine has emerged as another hydrophilic polymer option and is used in platforms such as PSARLink. Glucamine and glycan modifications are also being used to enhance solubility and stability.
However, hydrophilic modification must be carefully optimized. Excessive size or hydrophilicity may reduce tumor penetration or slow intracellular payload release. The best linker design must balance solubility, stability, enzymatic cleavage, tissue penetration, and payload potency.
Future Directions: Linkers as a Core Innovation Engine for ADCs
The future of ADC design will not be driven by antibodies or payloads alone. Linker engineering will remain one of the central innovation areas. Several directions are especially important.
First, researchers need better preclinical models that reflect human linker metabolism. Mouse plasma instability of certain linkers shows that animal models can misrepresent ADC stability and toxicity. Better cross-species assays and humanized models may improve translation.
Second, protease heterogeneity must be addressed. Tumors vary in cathepsin B, cathepsin L, legumain, elastase, and other protease expression. This means a linker that works well in one tumor type may not perform equally in another. Future ADC development may increasingly use protease biomarkers to guide linker selection.
Third, linker design should be integrated with antibody biology. Target internalization rate, lysosomal trafficking, antigen density, and tumor microenvironment features all influence whether a linker performs well. A linker cannot be optimized in isolation.
Finally, multifunctional linkers may become more common. Future ADCs may combine hydrophilic masking, exolinker architecture, tumor microenvironment activation, and highly specific cleavage sequences in one design. These advanced linkers may support dual-payload ADCs, degrader-antibody conjugates, immune-modulating ADCs, and non-oncology antibody conjugates.
Conclusion
Peptide linkers have moved from being a technical component of ADC chemistry to becoming a major driver of therapeutic performance. Classical linkers such as Val-Cit, Val-Ala, Ala-Ala, and GGFG have enabled important clinical advances, while new linker strategies are expanding what ADCs can achieve.
The next generation of peptide linkers is being designed to solve specific problems: premature cleavage, mouse plasma instability, neutrophil elastase sensitivity, aggregation, poor high-DAR compatibility, and limited tumor selectivity. Through sequence optimization, peptidomimetics, exolinker design, dual-cascade activation, and hydrophilic masking, researchers are building smarter ADCs with improved efficacy and safety.
For peptide and ADC researchers, the message is clear: linker design is no longer a supporting detail. It is a central determinant of ADC success. As precision oncology continues to evolve, peptide linkers will remain a key technology for turning targeted antibodies into safer, more powerful, and more adaptable cancer therapies.
Reference
Li, S., Guo, Y., Che, J., Dai, H., & Dong, X. (2025). Recent Advances in Peptide Linkers for Antibody-Drug Conjugates. Journal of Medicinal Chemistry, 68(19), 19871-19892.
https://doi.org/10.1021/acs.jmedchem.5c01982
Dumontet, C., Reichert, J. M., Senter, P. D., Lambert, J. M., & Beck, A. (2023). Antibody–drug conjugates come of age in oncology. Nature reviews Drug discovery, 22(8), 641-661.
https://doi.org/10.1038/s41573-023-00709-2
Walles, M., Connor, A., & Hainzl, D. (2017). ADME and safety aspects of non-cleavable linkers in drug discovery and development. Current topics in medicinal chemistry, 17(32), 3463-3475.
https://doi.org/10.2174/1568026618666180118153502
Ha, S. Y., Anami, Y., Yamazaki, C. M., Xiong, W., Haase, C. M., Olson, S. D., … & Tsuchikama, K. (2022). An enzymatically cleavable tripeptide linker for maximizing the therapeutic index of antibody–drug conjugates. Molecular cancer therapeutics, 21(9), 1449-1461.