Revolutionizing Peptide Synthesis: Fully Automated Azapeptide Integration in GLP-1 Design
Blog 212
Introduction: Automation Meets Peptide Chemistry
Peptide-based therapeutics have reshaped modern medicine, offering precise molecular tools for treating metabolic, cardiovascular, and neurological disorders. Molecules like GLP-1 analogues, including semaglutide, have demonstrated how well-designed peptides can combine potency with receptor selectivity. Yet their biggest limitation remains instability and enzymatic degradation. Each innovation in peptide chemistry seeks to extend lifespan and maintain bioactivity in biological systems.

Among the most promising advances are azapeptides, in which a nitrogen atom replaces the peptide backbone’s α-carbon, creating a more rigid and enzyme-resistant structure. Despite their potential, azapeptides have long been hindered by complex and inefficient synthesis. That barrier has finally been overcome. A new study from the Feinstein Institutes for Medical Research presents the first fully automated azapeptide synthesis platform, seamlessly integrating azapeptide construction into standard solid-phase peptide synthesis (SPPS) workflows.
The Hurdle: Complex Azapeptide Synthesis
For decades, chemists recognized azapeptides as valuable tools for enhancing peptide stability, yet their production was slow, hazardous, and difficult to automate. Traditional routes required activation of hydrazine derivatives using highly reactive reagents such as phosgene or chloroformates, which are toxic and unstable. These methods demanded strict manual control, low temperatures, and lengthy reaction times.
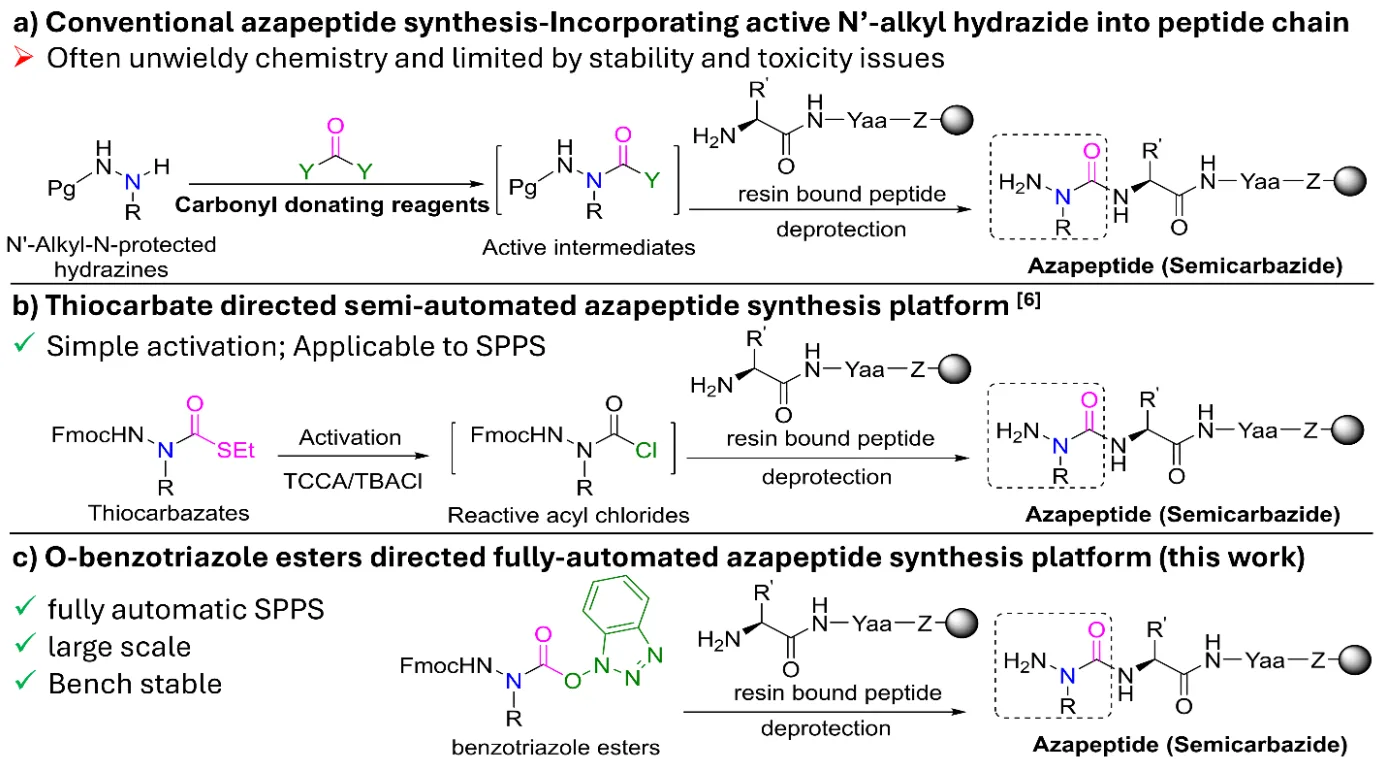
Even later improvements still suffered from low yields and poor compatibility with automated SPPS instruments. The problem wasn’t simply chemical—it was technological. Azapeptide coupling reactions often failed on standard synthesizers due to weak nucleophilicity of the modified backbone. Each new analogue required manual optimization, severely limiting innovation. Researchers needed a synthesis route that was bench-stable, reproducible, and SPPS-ready—laying the groundwork for the breakthrough that followed.
The Breakthrough: Fully Automated SPPS Integration
A research team led by Mingzhu He, Kai Fan Cheng, and Yousef Al-Abed introduced a true game-changer: a fully automated solid-phase azapeptide synthesis platform. At its core are Fmoc-protected benzotriazole ester building blocks (Fmoc-azaAA-OBt)—pre-activated, bench-stable aza-amino acids that integrate directly into conventional SPPS.
These building blocks maintain long-term stability, reacting smoothly with standard reagents and resins. The researchers successfully synthesized sixteen unique aza-amino acid monomers with 73–96% yields, all stable for months at room temperature. When incorporated into automated SPPS cycles, they achieved 85–100% conversion rates and excellent crude purities.
This innovation effectively transforms azapeptide synthesis into a plug-and-play process. Chemists can now build semicarbazide-containing peptides on the same instruments used for natural amino acid sequences—no custom chemistry or hazardous reagents required.
Optimization: Overcoming Coupling Challenges
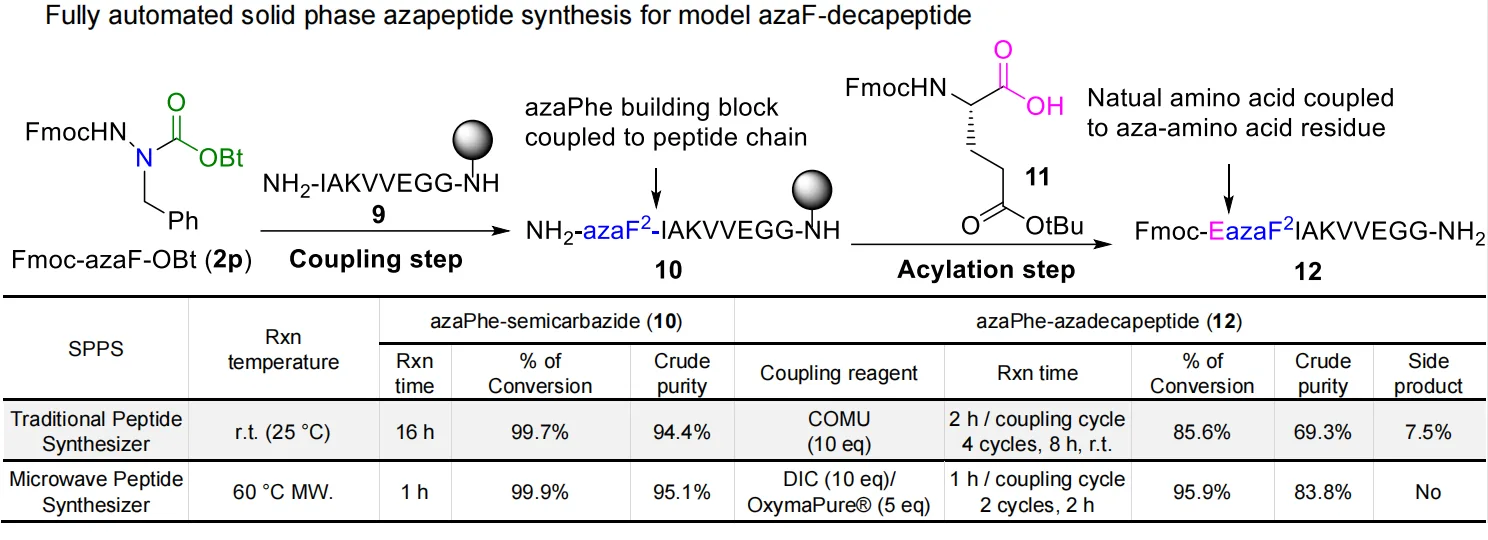
Despite automation, one challenge persisted: the semicarbazide nitrogen that forms after deprotection reacts sluggishly compared to the amine group in natural peptides. This reduced reactivity made extending the peptide chain difficult.
To solve it, the team systematically tested coupling reagents such as HCTU, COMU, DIC, and Oxyma Pure. The breakthrough came with microwave-assisted acylation at 60 °C using DIC/Oxyma Pure, which increased coupling efficiency to over 88%, halved the reaction time, and improved crude purity by minimizing side products.
This optimization step converted a slow, failure-prone reaction into a fast, reproducible process. By combining pre-activated aza-building blocks with microwave SPPS, the team established a universal method for constructing long and complex azapeptides efficiently—something previously considered impractical.
Case Study: Building GLP-1 Azapeptides
To demonstrate the system’s potential, the researchers synthesized analogues of glucagon-like peptide-1 (GLP-1)—a 31-residue hormone central to glucose regulation and insulin release. Native GLP-1 is degraded quickly by the enzyme DPP-IV, a challenge drug developers have long sought to overcome. Current treatments like semaglutide achieve stability by substituting unnatural amino acids such as Aib at the vulnerable site.
Using their automated method, the team replaced residues around the DPP-IV cleavage site with aza-alanine, aza-histidine, and aza-glutamic acid, generating four distinct GLP-1(7–37) analogues. These peptides were synthesized automatically within 4–6 hours for single substitutions and 7–11 hours for multi-aza variants—compared with more than 36 hours using traditional techniques.
The resulting products displayed 35–58% crude purity and 15–23% isolated yields. Notably, the [R³⁴, azaA⁸]-GLP-1(7–37) analogue achieved 22.4% yield and 96% purity, rivaling semaglutide’s Aib modification. This achievement marks the first fully automated synthesis of multi-aza GLP-1 analogues, proving that complex backbone-modified peptides can be made quickly and reproducibly.
Key Advantages of the Platform
The new synthesis system offers an unprecedented combination of speed, stability, and scalability:
Speed: Reaction times drop from days to hours, even for large peptides, thanks to microwave-assisted coupling and pre-activated esters.
Stability: Aza-amino acid substitutions create semicarbazide bonds that resist enzymatic cleavage, improving pharmacological durability.
Scalability: Fully compatible with standard Fmoc/tBu SPPS synthesizers, allowing rapid generation of azapeptide libraries without redesigning workflows.
Together, these features make it possible to produce enzyme-resistant analogues, receptor-binding variants, and backbone-modified peptide libraries at industrial efficiency. The method effectively bridges academic peptide design and large-scale pharmaceutical production.
Broader Impact on Peptide Drug Discovery
This technology represents a critical step toward next-generation peptide therapeutics. By automating azapeptide synthesis, researchers can now explore new chemical spaces that were previously inaccessible due to synthesis barriers. The resulting peptides combine the biological precision of natural sequences with the chemical robustness of small molecules.
Applications extend well beyond GLP-1. Azapeptide chemistry could strengthen candidates in cancer therapy, inflammation, infectious diseases, and immune regulation, where proteolytic degradation often undermines drug efficacy. The ability to generate diverse, custom azapeptide libraries also accelerates high-throughput screening and lead optimization.
In essence, this breakthrough merges automation and molecular engineering to create a pipeline for tailor-made, protease-resistant peptides, ushering in a new era of intelligent peptide drug design.
Conclusion: Toward Smarter Peptide Therapeutics
The advent of a fully automated azapeptide synthesis platform marks a pivotal milestone in peptide chemistry. By merging benzotriazole ester chemistry, microwave coupling, and SPPS automation, this method streamlines the creation of peptides that resist enzymatic degradation while preserving bioactivity.
As future studies explore stability, pharmacokinetics, and in vivo efficacy, this technology stands poised to reshape peptide drug development.
At LinkPeptide, we view this innovation as part of a larger transformation—where automation, chemistry, and biotechnology unite to deliver smarter, more resilient therapeutic peptides. The future of peptide innovation isn’t only about making molecules—it’s about making them better, faster, and more accessible to the scientific community.
Reference
He, M., Cheng, K. F., & Al-Abed, Y. (2025). Azapeptide Synthesis Fully Integrated into Standard Solid-Phase Peptide Synthesis (SPPS): Case Study with Azapeptide-GLP-1. bioRxiv, 2025-03.
https://doi.org/10.1101/2025.03.03.641232
Altiti, A., He, M., VanPatten, S., Cheng, K. F., Ahmed, U., Chiu, P. Y., … & Al-Abed, Y. (2022). Thiocarbazate building blocks enable the construction of azapeptides for rapid development of therapeutic candidates. Nature communications, 13(1), 7127.
https://doi.org/10.1038/s41467-022-34712-9
Tarchoun, K., Yousef, M. A., & Bánóczi, Z. (2022). Azapeptides as an Efficient Tool to Improve the Activity of Biologically Effective Peptides. Future Pharmacology, 2(3).
https://doi.org/10.3390/futurepharmacol2030020
Dinsmore, T. C., Liu, J., Miao, J., Ünsal, Ö., Sürmeli, D., Beinborn, M., … & Kumar, K. (2024). Potent and Protease Resistant Azapeptide Agonists of the GLP‐1 and GIP Receptors. Angewandte Chemie International Edition, 63(49), e202410237.