Sustainable Peptide Synthesis Meets Antimicrobial Power: A New Protocol for Hard-to-Make AMPs
Blog 153
Abstract
As antimicrobial resistance rises globally, antimicrobial peptides (AMPs) offer a powerful alternative to traditional antibiotics. However, synthesizing these peptides—especially long and hydrophobic ones—has been both environmentally taxing and technically challenging due to the limitations of standard Solid Phase Peptide Synthesis (SPPS). This blog explores a novel, sustainable approach that modifies the SPPS workflow using in situ Fmoc removal with pre-filtration, significantly reducing solvent waste and improving process efficiency. The method was successfully applied to synthesize three AMPs—P3, P4, and P5—derived from human SPLUNC1 and calgranulin C proteins. Among them, P3 demonstrated potent antimicrobial activity against E. coli and K. pneumoniae, comparable to colistin, without exhibiting cytotoxicity or hemolytic effects. This study showcases a green, efficient pathway to produce bioactive peptides with clinical potential, paving the way for safer and more sustainable therapeutic development in the era of resistant superbugs.
Old Methods, New Problems: Why Peptide Synthesis Needs a Green Makeover
Antimicrobial peptides (AMPs) are emerging as powerful tools in the fight against drug-resistant bacteria. These naturally occurring molecules, part of the body’s innate immune defense, can target a wide range of pathogens—including E. coli and Klebsiella pneumoniae—making them strong candidates for next-generation antibiotics.
However, turning these promising peptides into usable therapeutics presents a major hurdle: sustainability. The gold-standard method for peptide production, Solid Phase Peptide Synthesis (SPPS), consumes vast amounts of organic solvents. Over 80% of the waste in SPPS comes from repeated resin washing steps, contributing to alarmingly high Process Mass Index (PMI) and E-factor values.
The problem intensifies with hydrophobic peptides, which are notoriously difficult to synthesize due to aggregation and poor solubility. This leads to lower yields and even more solvent waste.
To meet the growing demand for peptide-based drugs, researchers are rethinking synthesis. In this article, we explore a modified SPPS protocol that dramatically reduces waste while enabling the efficient production of potent, hydrophobic antimicrobial peptides.
Breaking the Cycle: A Smarter, Cleaner Route to Complex Peptides
Traditional Solid Phase Peptide Synthesis (SPPS) follows a four-step cycle: coupling, washing, Fmoc deprotection, and another round of washing. While this method has been the backbone of peptide production for decades, its environmental footprint is hard to ignore. Solvent waste—especially from dimethylformamide (DMF) and dichloromethane (DCM)—dominates the process, often inflating the Process Mass Index (PMI) to more than 2000 for peptides longer than 10 amino acids.
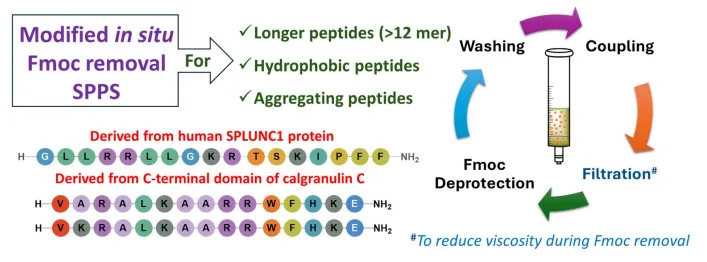
To address this, researchers have introduced a three-step protocol based on in situ Fmoc removal, which eliminates the need to wash the resin between coupling and deprotection. This not only reduces solvent use but also simplifies the workflow. Yet, even this approach faced limitations when applied to hydrophobic peptides, where viscous reaction mixtures and incomplete deprotection steps became problematic.
The latest breakthrough adds one crucial tweak: a filtration step before Fmoc removal. By removing the coupling cocktail prior to deprotection—especially after every fifth cycle—researchers were able to prevent aggregation and ensure efficient chain elongation. The method was tested in the synthesis of three antimicrobial peptides (P3, P4, and P5), all rich in hydrophobic and charged residues.
To further minimize waste, the protocol replaces multiple washes with just two quick rinses using 1% OxymaPure in DMF, a safer and more efficient reagent. The result? A significant reduction in PMI—down to 854–892—and improved synthesis of long, difficult peptides without compromising yield or purity.
This refined approach represents a meaningful step toward green peptide chemistry, particularly for complex sequences with therapeutic potential.
Nature-Inspired Designs: Building Powerful Peptides from Human Proteins
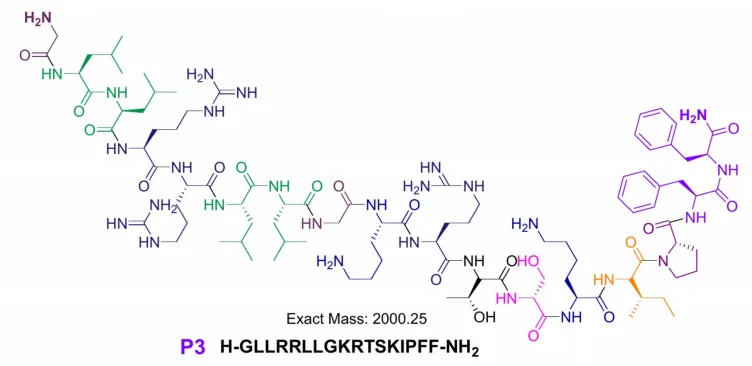
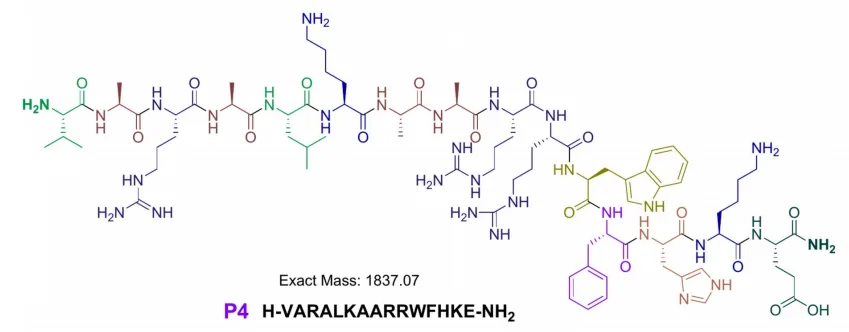
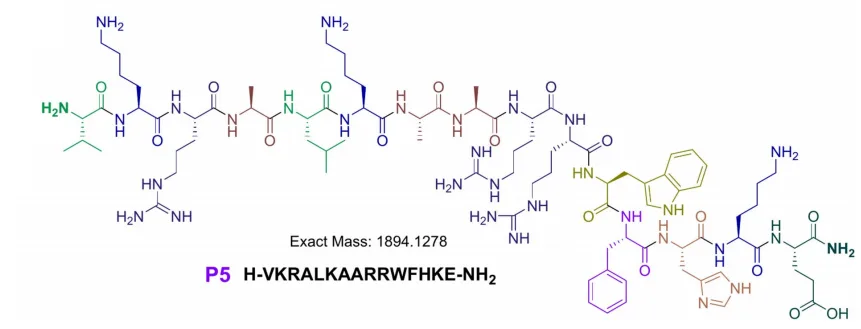
The proof of any synthesis method lies in the complexity and functionality of the peptides it can produce. In this study, the researchers targeted three newly designed antimicrobial peptides—P3, P4, and P5—inspired by the structure and function of natural human proteins.
Peptide P3 is derived from SPLUNC1 (Short Palate, Lung, and Nasal Epithelial Clone 1), a key component of the innate immune system known for its antimicrobial activity in the respiratory tract. P4 and P5 are based on the C-terminal domain of calgranulin C, a pro-inflammatory protein found in nasal and lung secretions.
What makes these peptides particularly challenging to synthesize is their high hydrophobicity—47% for P3 and P5, and 53% for P4. Hydrophobic amino acids tend to cause aggregation during synthesis, leading to poor yields and structural inconsistencies.
Despite these obstacles, all three peptides were successfully synthesized using the modified SPPS protocol, with yields exceeding 80%. Their purity and structure were confirmed by HPLC and MALDI-MS, validating the effectiveness of the streamlined method.
P3 in Action: Beating Superbugs with Eco-Friendly Peptides
Synthesis is just one side of the story—the true value of these peptides lies in their biological activity. To assess their therapeutic potential, peptides P3, P4, and P5 were tested against a panel of ESKAPE pathogens, a group of highly drug-resistant bacteria responsible for the majority of hospital infections. The tests also included Candida albicans, a common fungal pathogen.
The results were striking. Peptide P3 emerged as the most promising candidate, displaying potent antibacterial activity. It showed a minimum inhibitory concentration (MIC) of 6.25 µg/mL against E. coli—on par with the established antibiotic colistin—and retained effectiveness against Klebsiella pneumoniae with an MIC of 12.5 µg/mL.
Peptide P5 demonstrated moderate activity against K. pneumoniae (MIC = 25 µg/mL), while P4 showed no significant antimicrobial effect at tested concentrations.
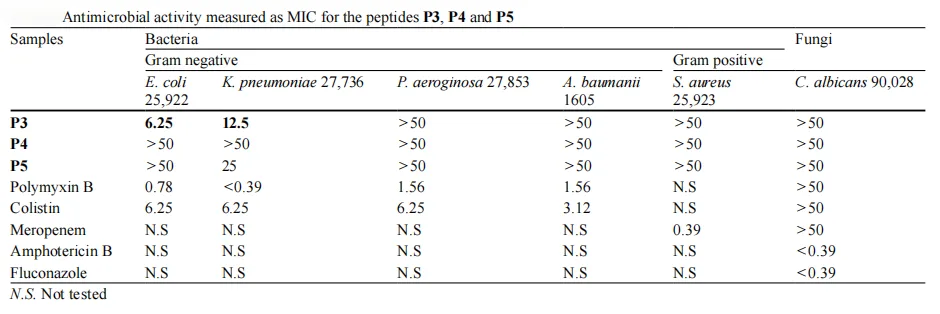
These findings highlight P3’s clinical potential as a lead antimicrobial peptide. Its potency against Gram-negative bacteria—combined with its sustainable synthesis—positions it as a strong candidate in the ongoing fight against multidrug-resistant infections.
Importantly, these bioactivity results were obtained without further peptide purification, underscoring both the robustness of the synthesis method and the functional integrity of the final products.
Safe, Strong, and Sustainable: Why These Peptides Check Every Box
In addition to antimicrobial potency, safety is a critical requirement for any therapeutic candidate. The researchers evaluated the peptides for cytotoxicity using human MCF-7 breast cancer cells and assessed hemolytic activity against red blood cells.
The results were highly encouraging. Even at concentrations as high as 200 µg/mL, all three peptides—P3, P4, and P5—exhibited no significant cytotoxic effects. Cell viability remained high across the board in MTT assays, suggesting excellent biocompatibility.
Hemolysis tests confirmed that the peptides were also non-toxic to red blood cells, with minimal release of hemoglobin compared to positive controls. This indicates that the peptides are safe for systemic exposure, a key consideration for antimicrobial agents targeting bloodstream infections.
Combined with the green synthesis strategy, these safety findings strengthen the case for advancing these peptides toward preclinical development. The study not only introduces a sustainable SPPS protocol, but also delivers functional, non-toxic AMPs that could meet the dual goals of environmental responsibility and medical effectiveness.
Reference
Anguraj, D., Chaudhary, S., Pasupuleti, M., Singh, L. R., & Sharma, A. (2025). Modified SPPS Protocol for the Synthesis of Novel Antimicrobial Peptides. International Journal of Peptide Research and Therapeutics, 31(4), 63.
https://doi.org/10.1007/s10989-025-10720-3
Al Musaimi, O., de la Torre, B. G., & Albericio, F. (2020). Greening Fmoc/t Bu solid-phase peptide synthesis. Green Chemistry, 22(4), 996-1018.
https://doi.org/10.1039/C9GC03982A
Rahangdale, R., Ghormode, P., Tender, T., Balireddy, S., Birangal, S., Kishore, R., … & Chandrashekar H, R. (2024). Anti-HSV activity of nectin-1-derived peptides targeting HSV gD: an in-silico and in-vitro approach. Journal of Biomolecular Structure and Dynamics, 1-14.
https://doi.org/10.1080/07391102.2024.2349525
Jadhav, S., Martin, V., Egelund, P. H., Castro, H. J., Krüger, T., Richner, F., … & Pedersen, D. S. (2021). Replacing DMF in solid-phase peptide synthesis: varying the composition of green binary solvent mixtures as a tool to mitigate common side-reactions. Green Chemistry, 23(9), 3312-3321.