Trofinetide: Advancing Therapeutic Horizons in Rett Syndrome and Neurodevelopmental Disorders
Blog 134
Abstract
Trofinetide, a synthetic analog of Glycine-Proline-Glutamate (GPE), represents a novel therapeutic approach in the treatment of Rett syndrome, a rare neurodevelopmental disorder characterized by significant cognitive and motor regression. Approved by the FDA in 2023, Trofinetide is the first treatment specifically designed for Rett syndrome, addressing a crucial unmet need. Through optimized pharmacodynamics and pharmacokinetics, Trofinetide enhances bioavailability and provides sustained neuroprotective effects. Clinical trials, including the pivotal LAVENDER Phase 3 study, demonstrated significant improvement in core symptoms, showing efficacy in both adolescent and pediatric populations. While Trofinetide presents a manageable safety profile, gastrointestinal effects like diarrhea are common and require monitoring. Further research is ongoing, with hopes to expand its indications and refine treatment protocols, reinforcing its potential in neurodevelopmental therapeutics. Trofinetide’s introduction marks a promising advancement for patients and researchers alike in addressing complex neurodevelopmental conditions.
Introduction to Trofinetide: A Novel Therapeutic for Neurodevelopmental Disorders
Trofinetide, marketed as DAYBUE™, represents a groundbreaking therapeutic advancement in the treatment of neurodevelopmental disorders, particularly Rett syndrome. Trofinetide is a synthetic analog of Glycine-Proline-Glutamate (GPE), the N-terminal tripeptide derivative of insulin-like growth factor-1 (IGF-1). This small molecule analog[1] is designed to harness the neurotrophic and neuroprotective properties of IGF-1, with a chemical structure optimized to cross the blood-brain barrier and facilitate greater bioavailability and a longer half-life than GPE itself.
Rett syndrome is a rare genetic disorder primarily affecting young females due to mutations in the MECP2 gene, which encodes a critical transcriptional regulator for neuronal function. This disorder is characterized by developmental regression, loss of motor and cognitive skills, and increased seizure susceptibility, among other complex symptoms[2]. Trofinetide’s therapeutic potential in Rett syndrome is particularly significant, as treatment options have historically been limited to symptomatic management.
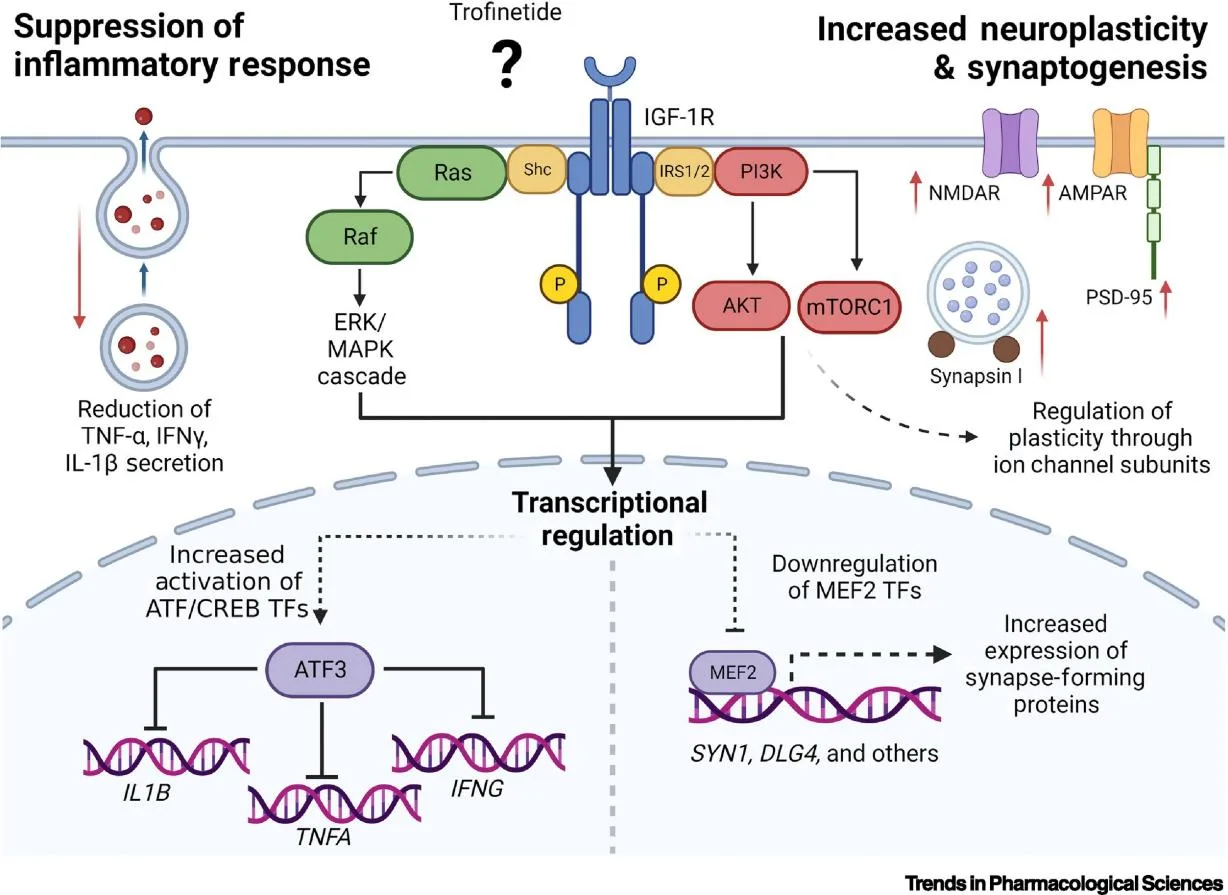
While the precise mechanism by which Trofinetide exerts its effects in Rett syndrome remains unknown, preclinical studies have shown that administration of GPE analogs can partially restore dendritic spine density, enhance synaptic plasticity, and stabilize neurological function in Rett syndrome models[3]. These findings highlight Trofinetide’s potential role in modifying the underlying neuropathology of this challenging disorder.
Pharmacodynamics and Pharmacokinetics of Trofinetide: Optimized for Efficacy
Trofinetide’s pharmacodynamic and pharmacokinetic profiles highlight its enhanced potential for therapeutic efficacy in Rett syndrome compared to its precursor, GPE. Structurally, Trofinetide is methylated at the 2-position of proline, which significantly increases its oral bioavailability and extends its elimination half-life. This optimization allows Trofinetide[4] to maintain stable plasma levels, enhancing its capacity to penetrate the blood-brain barrier and produce sustained neuroprotective effects in the central nervous system.
The pharmacokinetics of Trofinetide indicate a consistent and linear absorption profile, with peak plasma concentrations occurring approximately 2 to 3 hours after administration. Studies demonstrate that systemic exposure to Trofinetide is dose-proportional across a range of tested doses, and repeated administration does not lead to significant accumulation, supporting its safety in long-term use. Additionally, Trofinetide’s metabolism is minimal, with low interaction potential with hepatic CYP450 enzymes, a factor that reduces concerns about drug-drug interactions and makes it a promising candidate for combination therapies.
As most of the administered dose is excreted unchanged in urine, Trofinetide’s clearance pathway is relatively predictable, with fecal elimination being a minor route. Co-administration with high-fat meals decreases peak plasma concentration by around 20%, but does not impact overall drug exposure (AUC). This allows for some flexibility in administration, as patients can take the medication with or without food. These pharmacokinetic properties make Trofinetide a well-suited option for chronic use in managing Rett syndrome’s complex symptomatology.
Clinical Trials and Efficacy: Trofinetide’s Impact on Rett Syndrome Symptoms
Trofinetide has demonstrated promising efficacy in clinical trials for Rett syndrome, with several studies underscoring its impact on core symptoms. The LAVENDER Phase 3 trial, a pivotal study involving girls and women aged 5 to 20 years with Rett syndrome, showed significant improvements in key outcome measures, including the Rett Syndrome Behavior Questionnaire (RSBQ) and Clinical Global Impression-Improvement (CGI-I) scores[5]. Trofinetide-treated patients experienced a greater reduction in RSBQ total scores and showed improvements in CGI-I ratings compared to those on placebo, suggesting meaningful enhancements in symptom severity and global functioning.
The DAFFODIL Phase 2/3 trial further explored Trofinetide’s safety and efficacy in younger children, aged 2 to 4 years, with Rett syndrome. This open-label study indicated improvements in the Caregiver Global Impression–Improvement (CGI-I) and quality-of-life metrics, confirming the drug’s benefits in early stages of the disease[6]. Moreover, the LILAC extension study, which followed participants from the LAVENDER trial, demonstrated sustained symptom improvement and safety over 40 weeks of treatment. In LILAC, patients who switched from placebo to Trofinetide[7] continued to show positive response trends, providing evidence for the drug’s long-term potential in managing Rett syndrome.
Collectively, these trials establish Trofinetide as an effective option for alleviating the core behavioral and cognitive challenges of Rett syndrome, supporting its unique value in addressing this unmet medical need.
Safety Profile and Adverse Events: Managing Side Effects of Trofinetide
Trofinetide’s safety profile has been extensively studied, with clinical trials providing insights into the most common adverse events and strategies for managing them. In the Phase 3 LAVENDER trial, diarrhea was reported as the most frequent treatment-emergent adverse event, affecting approximately 82% of patients on Trofinetide compared to 20% in the placebo group. While generally mild to moderate in severity, diarrhea led to dose adjustments, interruptions, or, in some cases, discontinuation of treatment. Most cases were managed with antidiarrheal medications, emphasizing the need for proactive monitoring.
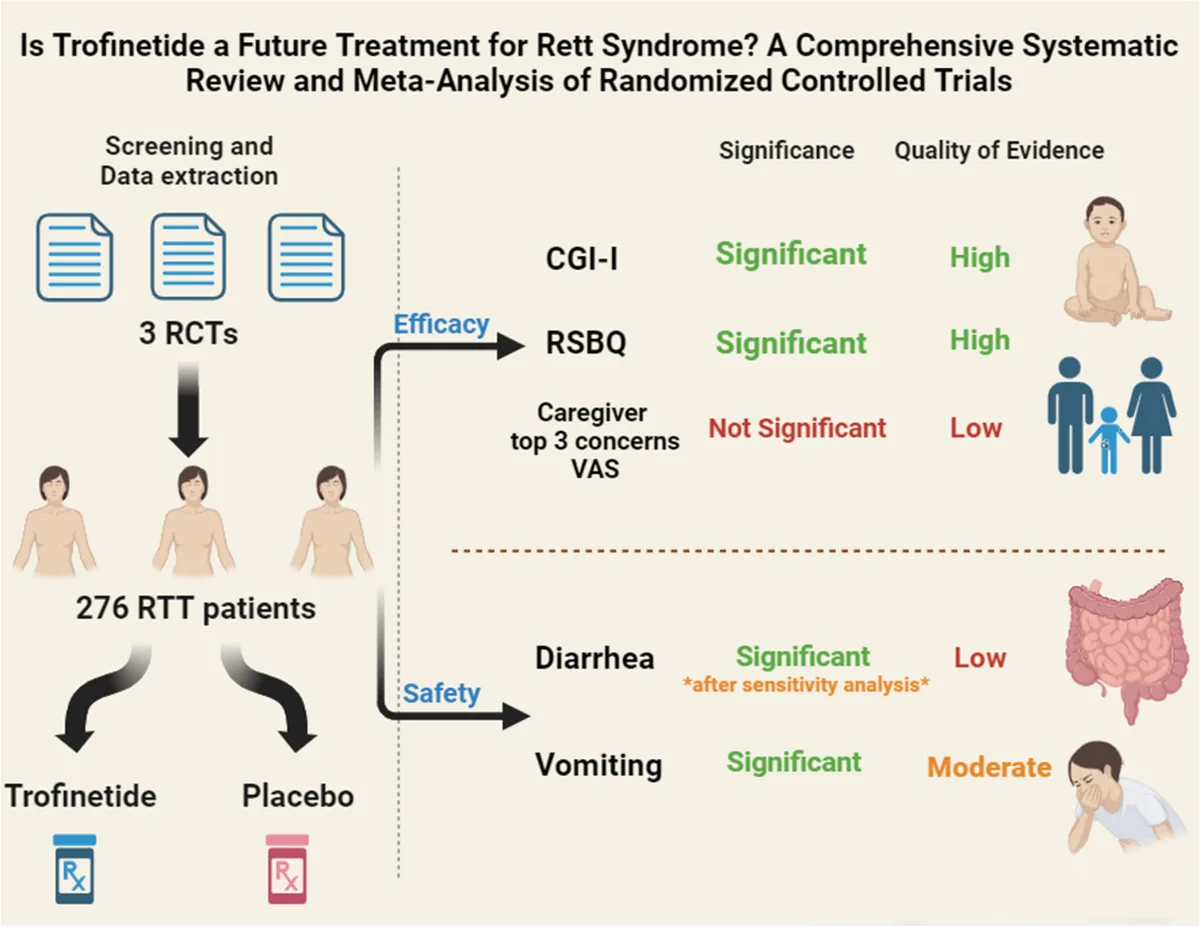
Vomiting was another common side effect, observed in 29% of Trofinetide-treated patients versus 12% on placebo. Other reported adverse events included fatigue, decreased appetite, and fever, though these were generally mild and manageable. Notably, some patients experienced significant weight loss (>7% from baseline), prompting additional guidance on monitoring body weight and adjusting treatment as needed. In long-term studies, such as the LILAC extension trial, these adverse events were consistent, further affirming Trofinetide’s safety profile over extended use.
In younger children aged 2 to 4 years participating in the DAFFODIL trial, similar safety outcomes were reported, with diarrhea and vomiting being the primary adverse events[6]. The data indicate that while Trofinetide’s side effects are manageable, vigilant monitoring for gastrointestinal symptoms and weight changes is essential for optimizing treatment outcomes in patients with Rett syndrome.
Regulatory Milestones and Future Directions: Expanding Trofinetide’s Therapeutic Potential
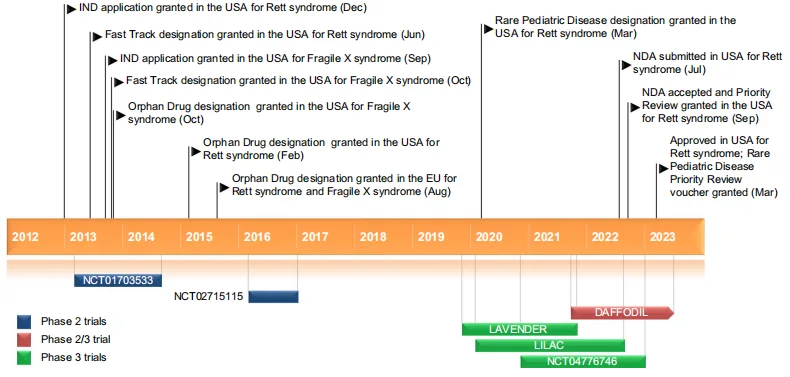
Trofinetide’s recent regulatory milestones and ongoing research emphasize its potential as a novel therapeutic in treating neurodevelopmental disorders. In March 2023, the FDA granted Trofinetide its first approval for treating Rett syndrome in patients aged two years and older. This approval marks a significant achievement, as Trofinetide is the first drug authorized specifically for Rett syndrome, a condition with limited treatment options. The drug’s regulatory pathway also benefited from priority review and orphan drug status, highlighting its value in addressing a rare and underserved medical condition.
The licensing agreement between Neuren Pharmaceuticals and Acadia Pharmaceuticals has also facilitated Trofinetide’s commercialization across North America, ensuring greater availability for patients in need. Neuren retains the rights for global markets, supporting potential future approvals beyond the United States. Additionally, Trofinetide has shown promise in treating other neurodevelopmental conditions, with research extending to Fragile X syndrome. Although early trials have shown some efficacy in addressing cognitive and behavioral symptoms in Fragile X, further studies are needed to validate these results and expand its indications.
Ongoing clinical trials, such as the DAFFODIL study in young children and the LILAC extension study, continue to assess Trofinetide’s long-term safety and efficacy. These studies will likely contribute valuable data for refining treatment protocols and expanding therapeutic applications, reinforcing Trofinetide’s role as a pioneering option for neurodevelopmental disorders.
Reference
- Keam, S. J. (2023). Trofinetide: first approval. Drugs, 83(9), 819-824.
- Collins, B. E., & Neul, J. L. (2022). Rett syndrome and MECP2 duplication syndrome: disorders of MeCP2 dosage. Neuropsychiatric Disease and Treatment, 2813-2835.
- Tropea, D., Giacometti, E., Wilson, N. R., Beard, C., McCurry, C., Fu, D. D., … & Sur, M. (2009). Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proceedings of the National Academy of Sciences, 106(6), 2029-2034.
- Darwish, M., Youakim, J. M., Harlick, J., DeKarske, D., & Stankovic, S. (2022). A phase 1, open-label study to evaluate the effects of food and evening dosing on the pharmacokinetics of oral trofinetide in healthy adult subjects. Clinical Drug Investigation, 42(6), 513-524.
- Glaze, D. G., Neul, J. L., Kaufmann, W. E., Berry-Kravis, E., Condon, S., Stoms, G., … & Rett 002 Study Group. (2019). Double-blind, randomized, placebo-controlled study of trofinetide in pediatric Rett syndrome. Neurology, 92(16), e1912-e1925.
- Percy, A., Ryther, R., Marsh, E., Feyma, T., Lieberman, D., Neul, J., … & Youakim, J. (2023). Trofinetide for the treatment of Rett syndrome: an open-label study in girls 2 to 4 years of age (P13-9.005). Neurology, 100(17_supplement_2), 1378.
- Percy, A. K., Neul, J. L., Benke, T. A., Berry-Kravis, E. M., Glaze, D. G., Marsh, E. D., … & Youakim, J. M. (2024). Trofinetide for the treatment of Rett syndrome: results from the open-label extension LILAC study. Med, 5(9), 1178-1189.