Unlocking the Undruggable: CPPTACs and the Future of Membrane Protein Therapies
Blog 345
Abstract
Targeted protein degradation (TPD) has revolutionized modern drug discovery, yet plasma membrane proteins—critical in cancer, immunity, and metabolism—have remained stubbornly difficult to degrade. Traditional PROTACs act only on intracellular targets, while lysosome-based systems like LYTACs depend on tissue-specific receptors and suffer from efficiency limitations. A recent Nature Communications study introduces a novel solution: Cell-Penetrating Peptide–Targeting Chimeras (CPPTACs). By linking cell-penetrating peptides to small-molecule binders, CPPTACs drive endocytosis and lysosomal degradation of membrane proteins in a receptor-independent manner. Proof-of-concept studies highlight efficient degradation of PD-L1, CAIX, and CB2R, demonstrating broad applicability across immune checkpoints, tumor metabolism, and GPCR signaling. Unlike earlier platforms, CPPTACs avoid the hook effect, maintain a wide therapeutic window, and are simple to design. While challenges remain in peptide stability and pharmacokinetics, CPPTACs represent a paradigm shift that extends TPD to previously undruggable protein classes with exciting clinical potential.
Breaking Barriers: Why Membrane Proteins Are Hard to Degrade
Cell surface proteins are the gatekeepers of biology. From receptors and transporters to immune checkpoints, they orchestrate how cells sense their surroundings, communicate with neighbors, and respond to stress or disease. Their accessibility makes them some of the most attractive drug targets in medicine, driving therapies that reshape cancer treatment, metabolic control, and immunotherapy.
But when it comes to selective degradation, membrane proteins have long resisted modern tools. The rise of targeted protein degradation (TPD) transformed drug discovery with PROTACs (Proteolysis-Targeting Chimeras), which recruit the proteasome to remove harmful proteins inside the cell. These compounds offer unmatched precision for cytosolic or nuclear targets. Yet PROTACs simply cannot reach proteins embedded in the plasma membrane, leaving a blind spot in the TPD revolution.
To bridge this gap, researchers have developed lysosome-based degraders such as LYTACs (Lysosome-Targeting Chimeras). These molecules tether surface proteins to lysosomal receptors, funneling them into the cell’s waste system. The concept is powerful, but the execution is uneven: lysosome-targeting receptors vary between tissues, limiting where LYTACs work. Even when effective, they often suffer from the so-called “hook effect,” where higher drug concentrations paradoxically reduce degradation efficiency.
This challenge has kept many clinically important surface proteins out of reach. How do we design a strategy that works independently of receptor expression, avoids efficiency pitfalls, and remains broadly applicable across cell types? A recent study published in Nature Communications introduces a promising answer: a deceptively simple yet powerful platform that uses cell-penetrating peptides (CPPs) to drive membrane protein degradation in a universal way.
Introducing CPPTACs: Harnessing Cell-Penetrating Peptides for Degradation
The new platform is built on a simple idea: harness cell-penetrating peptides (CPPs) as universal drivers of endocytosis. CPPs are short, positively charged peptide sequences—like PEN, TAT, or R9—that can slip across cell membranes and carry molecular cargo with them. For years, they have been explored as delivery vehicles for proteins, nucleic acids, and drugs. But their tendency to get trapped inside endosomes, often seen as a drawback, can be turned into an advantage.
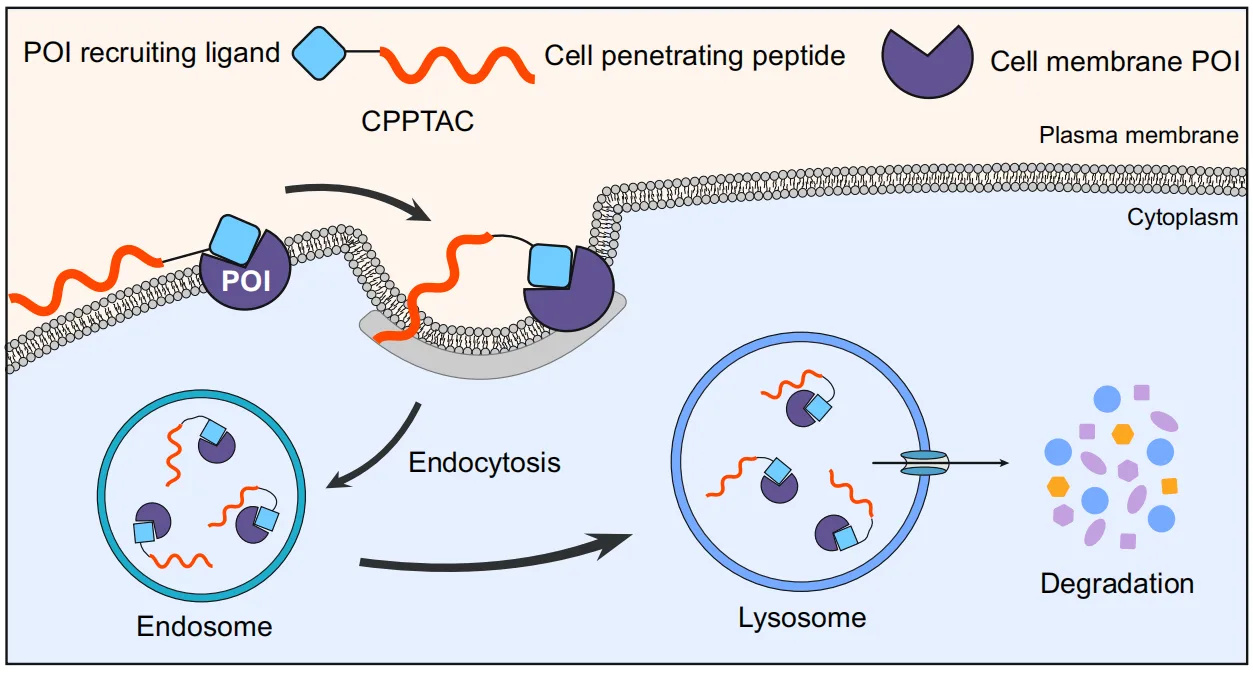
By chemically linking CPPs to small-molecule ligands that bind specific membrane proteins, the researchers created CPP-mediated lysosome-targeting chimeras (CPPTACs). Once the small molecule attaches to its target, the CPP component pulls the complex into the cell through endocytosis. Instead of escaping into the cytosol, the internalized protein–CPPTAC complex naturally traffics to lysosomes, where it is degraded.
This approach bypasses the limitations of LYTACs and similar technologies. Because CPPs can induce endocytosis without relying on lysosome-targeting receptors, CPPTACs work across different cell types, regardless of receptor expression levels. Their design is also streamlined: only two components—a peptide and a ligand—are required, unlike the three-part binding model of other degraders. Importantly, CPPTACs do not suffer from the “hook effect”, giving them a broader and safer therapeutic window.

In early tests, four different CPPs were evaluated, with the PEN and TAT sequences emerging as the most effective at driving protein internalization and degradation. This flexibility suggests that CPPTACs can be tailored to diverse biological contexts, making them a versatile addition to the protein degradation toolbox.
Proof in Action: How CPPTACs Tackle PD-L1, CAIX, and CB2R
To test whether CPPTACs could truly deliver on their promise, the researchers applied the platform to three clinically significant membrane proteins, each representing a different therapeutic frontier.
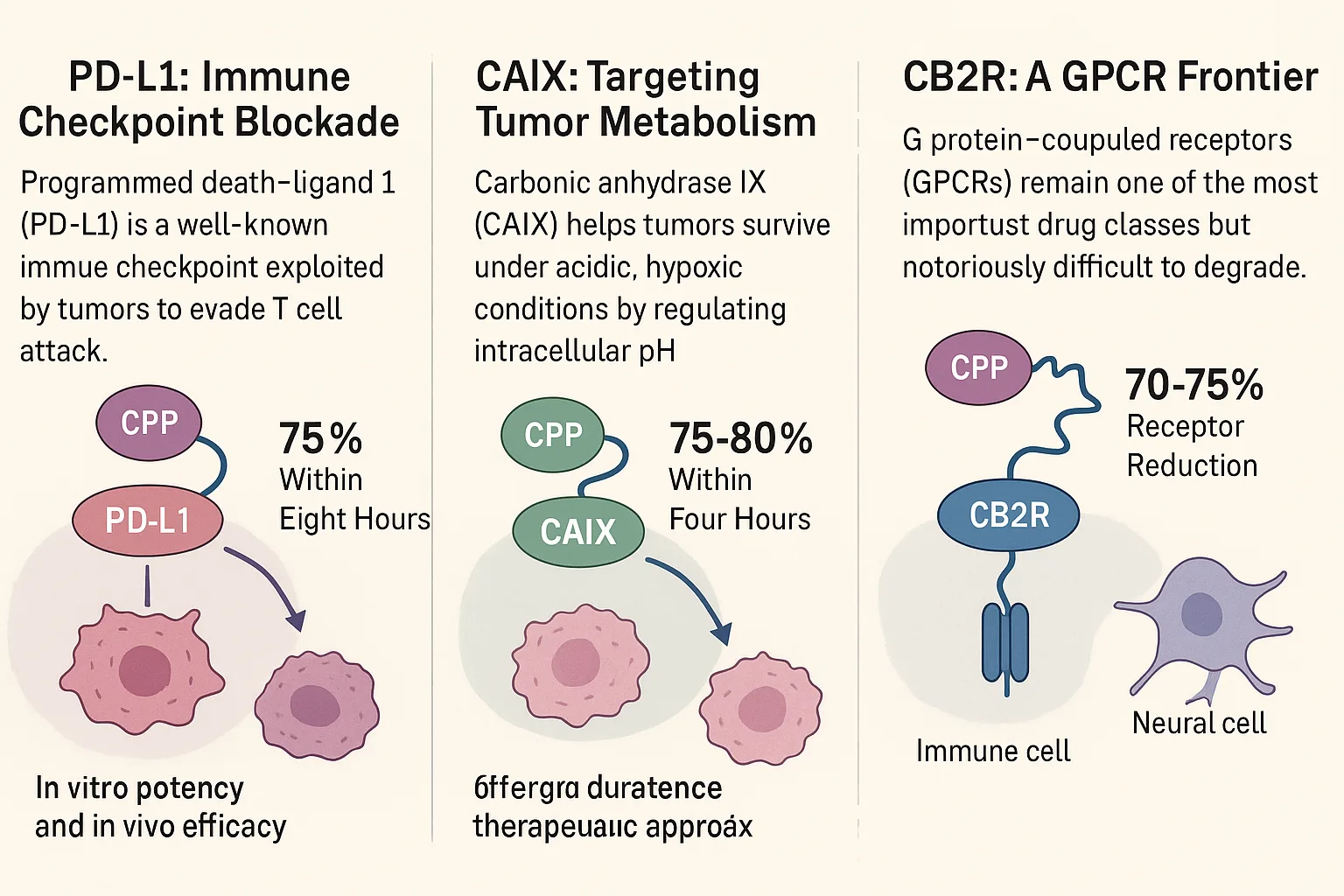
1. PD-L1: Immune Checkpoint Blockade
Programmed death-ligand 1 (PD-L1) is a well-known immune checkpoint exploited by tumors to evade T cell attack. By conjugating a PD-L1–binding small molecule (BMS-8) to CPPs, the team developed BMS-CPP1 and BMS-CPP2. In cancer cells, these conjugates achieved rapid PD-L1 degradation—up to 75% within eight hours—without the dose-limiting hook effect. In mouse tumor models, BMS-CPP1 suppressed tumor growth, reduced metastasis, and decreased PD-L1 levels in tumor tissue, demonstrating both in vitro potency and in vivo efficacy.
2. CAIX: Targeting Tumor Metabolism
Carbonic anhydrase IX (CAIX) helps tumors survive under acidic, hypoxic conditions by regulating intracellular pH. It is strongly upregulated in aggressive cancers. CPPTACs built from a CAIX-binding small molecule (Sul-CPP1 and Sul-CPP2) induced 75–80% degradation of CAIX within four hours, even under hypoxic stress. Treated cancer cells showed dramatically reduced survival, rivaling the effect of SLC-0111, a CAIX inhibitor in clinical trials. Unlike inhibitors, however, CPPTACs removed the protein entirely, offering a more durable therapeutic approach.
3. CB2R: A GPCR Frontier
G protein–coupled receptors (GPCRs) remain one of the most important drug classes but are notoriously difficult to degrade. Using HU-CPP1, a conjugate targeting the cannabinoid type 2 receptor (CB2R), the researchers achieved 70–75% receptor reduction across multiple cell types, including immune and neural models. This result highlights CPPTACs’ capacity to tackle challenging seven-transmembrane proteins, opening opportunities in inflammation and neurodegeneration research.
Together, these proof-of-concept studies establish CPPTACs as a versatile, broadly applicable platform. Whether the target is an immune checkpoint, a metabolic enzyme, or a GPCR, CPPTACs can induce efficient lysosomal degradation, expanding the landscape of druggable proteins.
Why CPPTACs Stand Out: Advantages Over Existing Approaches
The success of CPPTACs against PD-L1, CAIX, and CB2R demonstrates more than just proof-of-concept—it highlights a fundamentally new way to expand the therapeutic reach of targeted protein degradation. Unlike receptor-dependent systems such as LYTACs, CPPTACs harness the inherent ability of cell-penetrating peptides to drive endocytosis. This receptor independence means they can, in principle, function across many cell types and tissues without being constrained by lysosome-targeting receptor expression.
Another key advantage is the absence of the hook effect, which has long limited the effectiveness of classical bifunctional degraders. Because CPPTACs rely on straightforward peptide–protein and ligand–target interactions, they maintain activity across a broader concentration range. This translates to a wider therapeutic window, an essential consideration in drug development where dose flexibility can reduce toxicity risks.
The design of CPPTACs is also refreshingly simple. Each construct consists of only two components: a small molecule that recognizes the protein of interest and a peptide that ensures cellular uptake and lysosomal delivery. This minimalism not only makes them easier to synthesize but also more adaptable to different disease targets.
From a translational perspective, CPPTACs hold promise in diverse therapeutic arenas:
- Cancer immunotherapy, by depleting PD-L1 and restoring immune surveillance.
- Tumor metabolism, by removing CAIX and weakening cancer survival in hypoxic niches.
- Neurological and inflammatory diseases, by modulating CB2R signaling.
By combining the versatility of small molecules with the trafficking power of CPPs, CPPTACs emerge as a platform with broad potential for both research applications and future clinical therapies.
From Concept to Clinic: Challenges and What Comes Next
While CPPTACs offer a compelling new strategy, important challenges remain before they can be fully translated into the clinic. A major hurdle is the nature of cell-penetrating peptides themselves. Although effective at inducing endocytosis, CPPs can suffer from poor metabolic stability, rapid clearance, or unintended interactions at high doses. Addressing these issues will be critical to ensure safety and durability in patients.
Strategies are already on the horizon. Cyclization of peptides, incorporation of non-natural amino acids, or the design of prodrug forms may improve pharmacokinetics and reduce potential cytotoxicity. Beyond chemistry, tailoring CPPTACs to different disease contexts—such as solid tumors, autoimmune disorders, or neurodegenerative conditions—will require careful optimization of both the ligand and peptide components.
Looking ahead, the promise of CPPTACs lies in their universality. By offering a receptor-independent and broadly applicable method to degrade membrane proteins, they extend the reach of targeted protein degradation into domains previously considered undruggable. As refinements improve stability and selectivity, CPPTACs may well emerge as a next-generation therapeutic platform—bridging the gap between basic research and transformative clinical applications.
Reference
He, W., Chen, C., Zheng, J., Li, Y., Shi, H., Zhou, Y., … & Fang, L. (2025). Targeted degradation of cell surface proteins through endocytosis triggered by cell-penetrating peptide-small molecule conjugates. Nature Communications, 16(1), 7575.
https://doi.org/10.1038/s41467-025-62776-w
Alabi, S. B., & Crews, C. M. (2021). Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. Journal of Biological Chemistry, 296.
https://doi.org/10.1016/j.jbc.2021.100647
Békés, M., Langley, D. R., & Crews, C. M. (2022). PROTAC targeted protein degraders: the past is prologue. Nature reviews Drug discovery, 21(3), 181-200.
https://doi.org/10.1038/s41573-021-00371-6
Zhou, Y., Teng, P., Montgomery, N. T., Li, X., & Tang, W. (2021). Development of triantennary N-acetylgalactosamine conjugates as degraders for extracellular proteins. ACS Central Science, 7(3), 499-506.